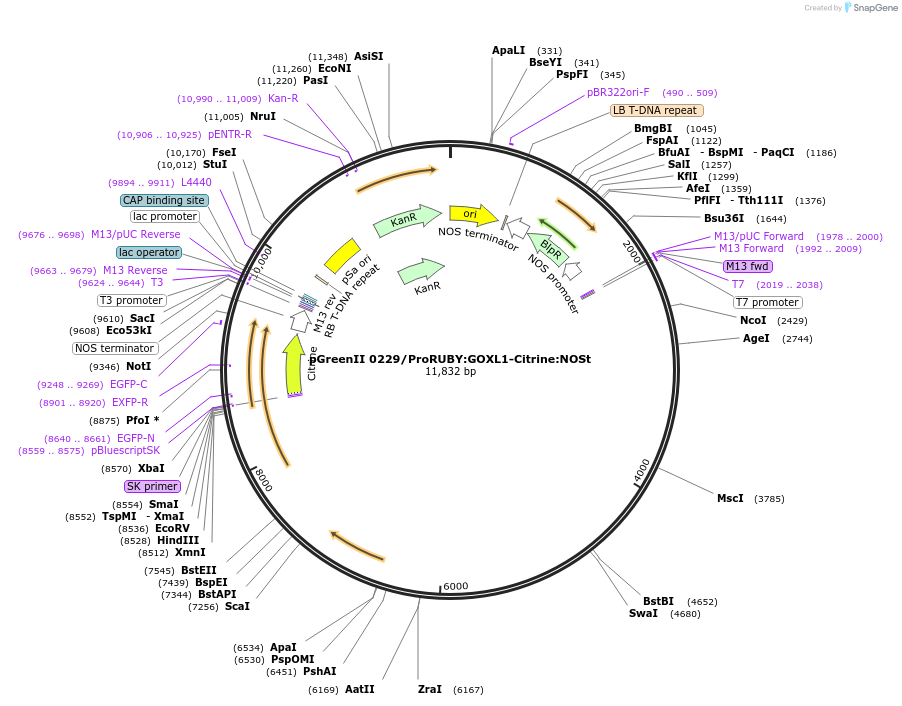
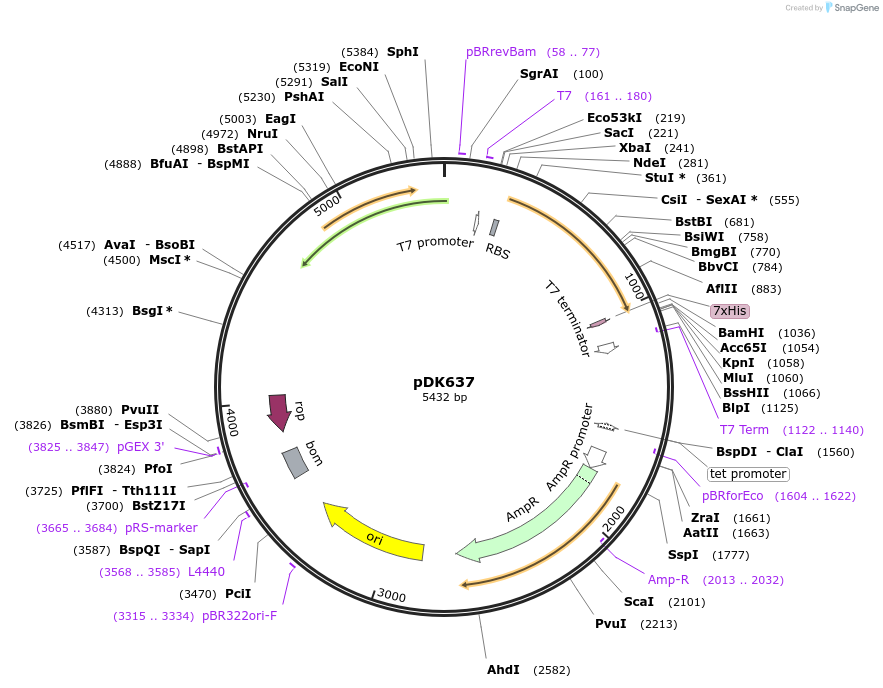
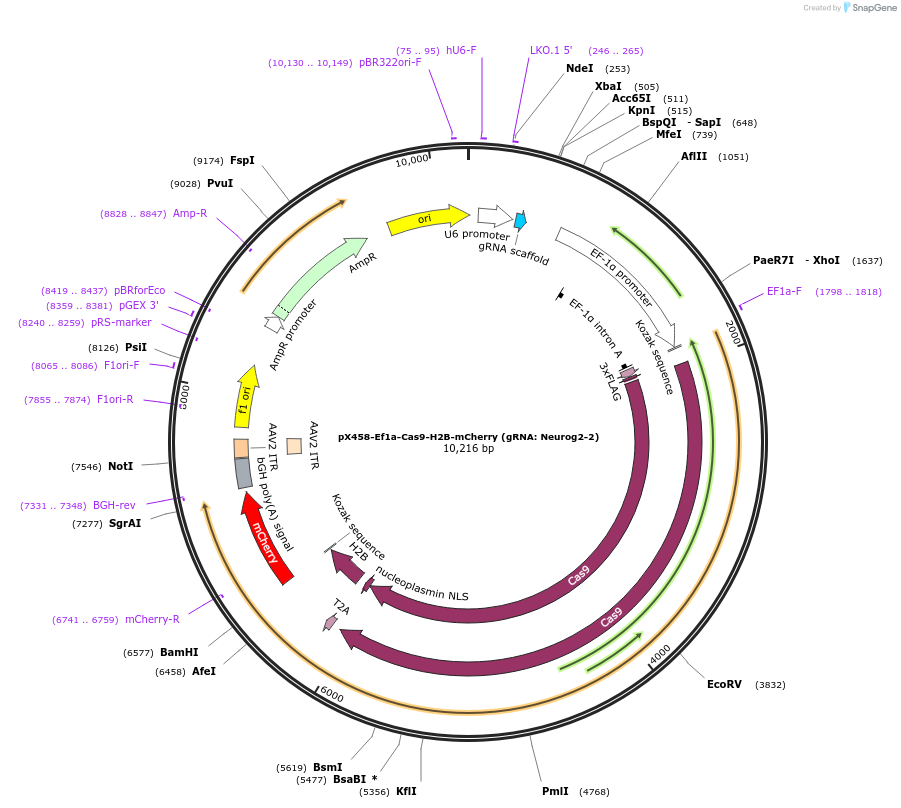
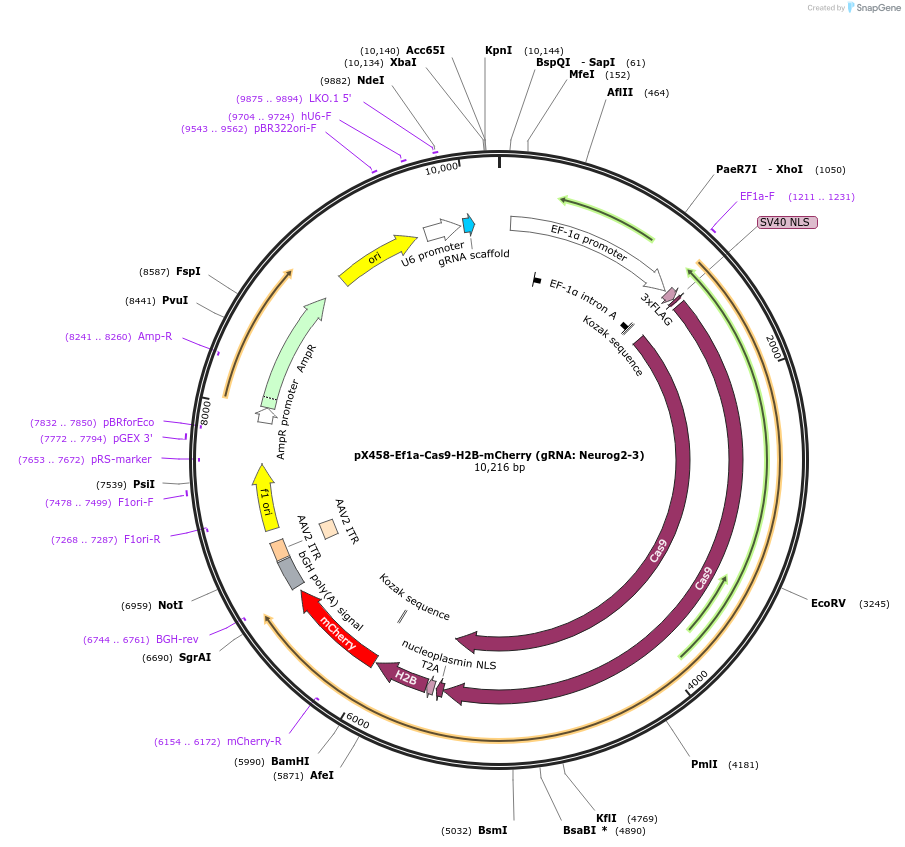
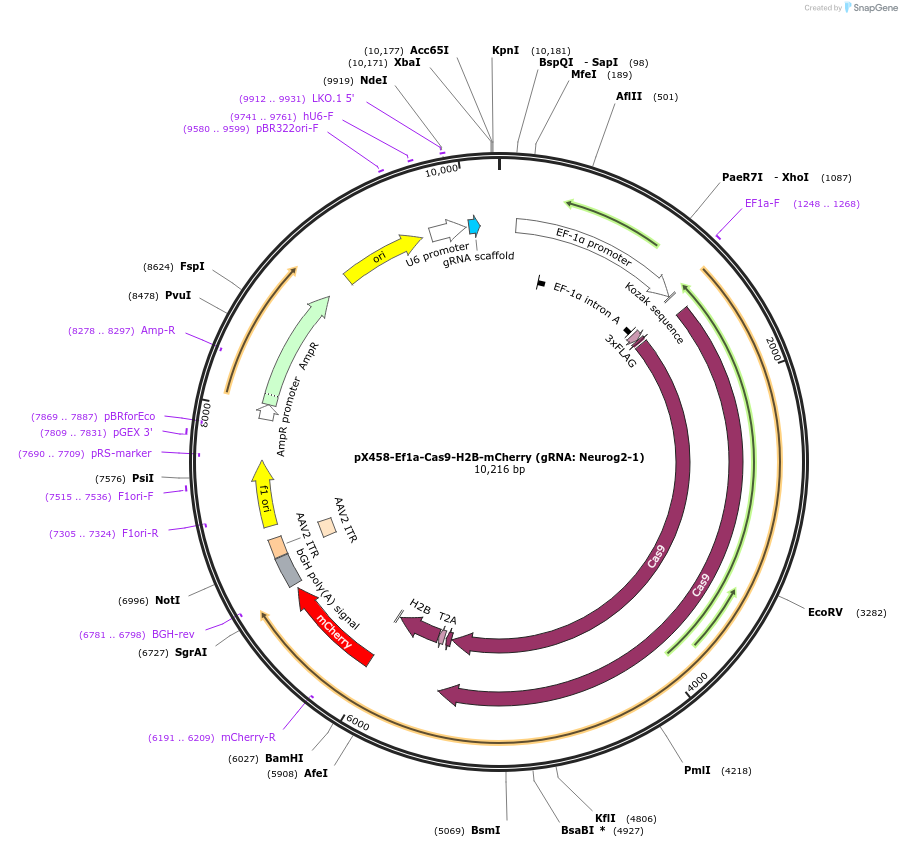
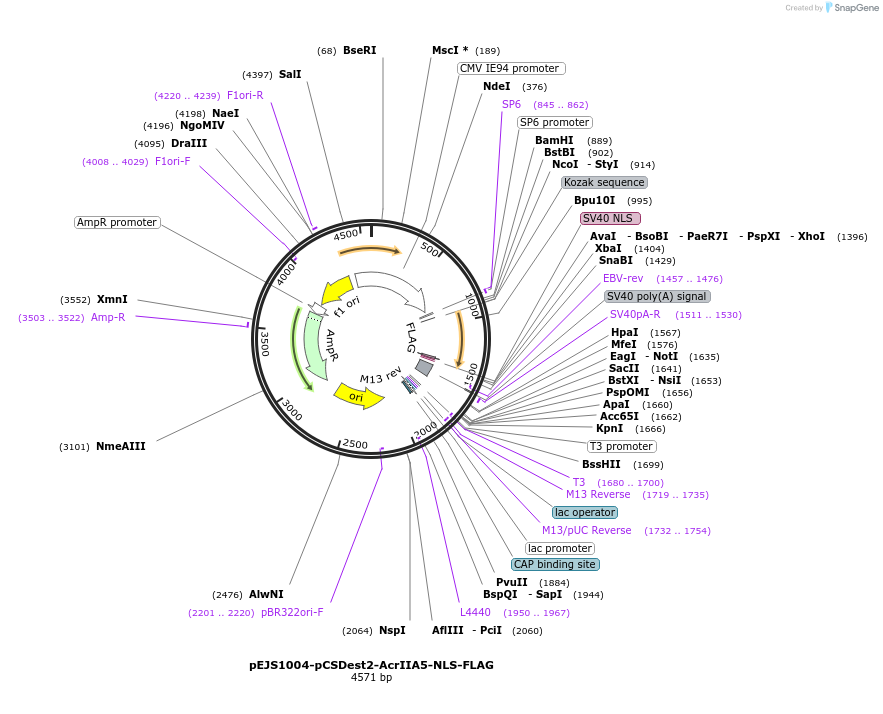
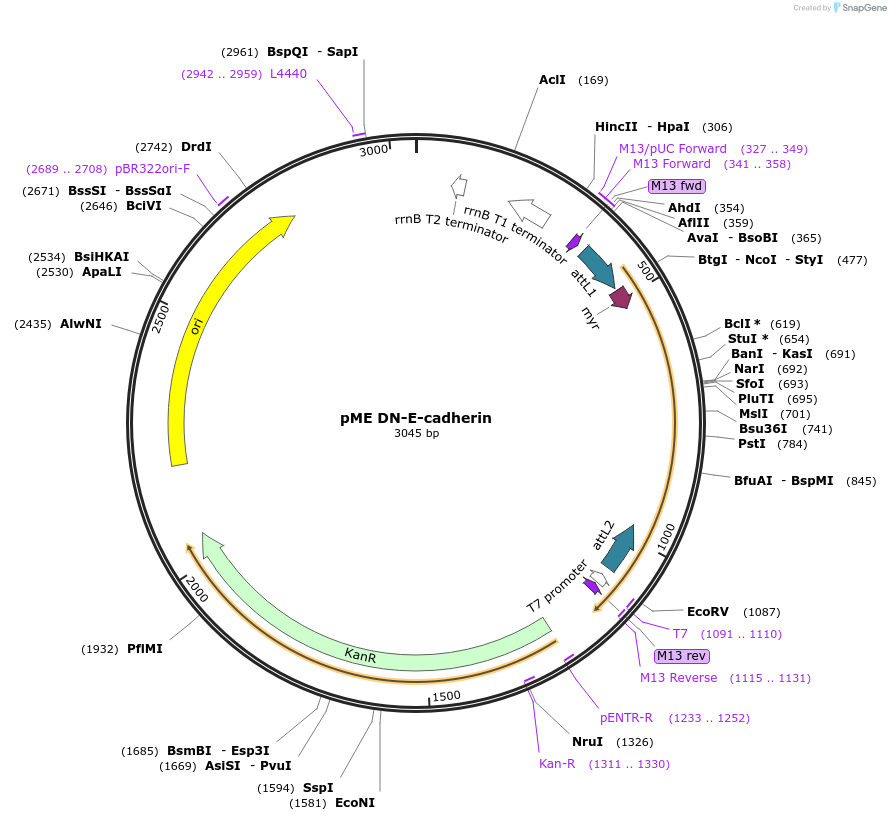
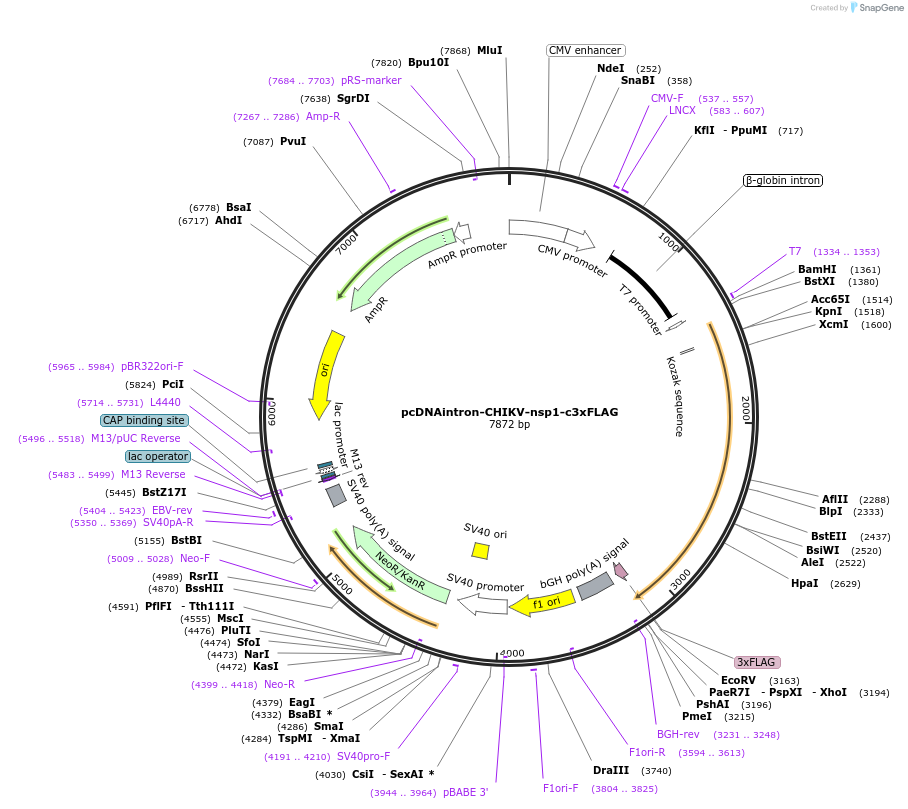
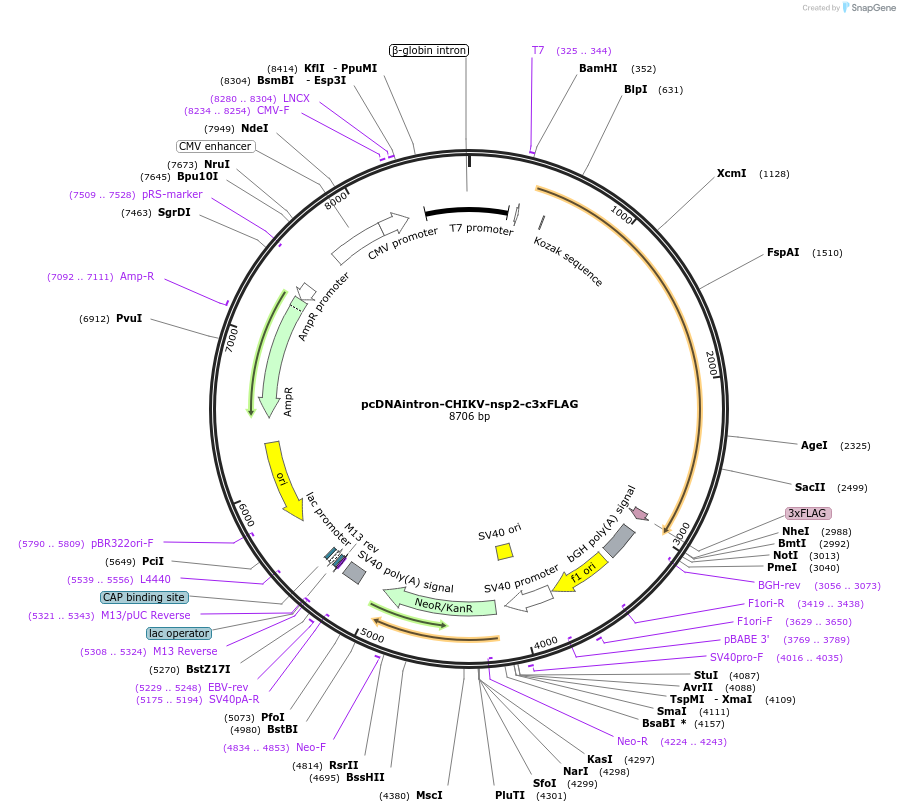
pGreenII 0229/ProRUBY:GOXL1-Citrine:NOSt
Plasmid
#175563
Purpose
pSOUP helper plasmid needed for Agrobacterium-mediated plant transformation - Expression of GOXL genes tagged with Citrine under RUBY promoter for transgene complementation of ruby-1
Depositor
Insert
GOXL1
Tags
Citrine
Expression
Plant
Promoter
RUBY
Available Since
Oct. 19, 2021
Availability
Academic Institutions and Nonprofits only