
Molecular Biology Reference
Plasmids are one of the most important tools in molecular biology. They allow scientists to isolate specific genes for study and have led to the understanding of many important molecular pathways and diseases. Here we present an introduction to plasmids and a collection of other molecular biology resources, including:
Origins of Molecular Genetics
The concept of genes as carriers of phenotypic information was introduced in the early 19th century by Gregor Mendel, who later demonstrated the properties of genetic inheritance in peas. Over the next 100 years, many significant discoveries led to the conclusion that genes encode proteins and reside on chromosomes, which are composed of DNA. These findings culminated in the central dogma of molecular biology, that proteins are translated from RNA, which is transcribed from DNA.
The Genetic Code
The genetic code is a set of rules for translating the information encoded by DNA and RNA into proteins. DNA is comprised of four nucleotide bases: adenine, thymine, cytosine, and guanine (abbreviated to A, T, C, and G, respectively). The bases are organized into a right-handed, double-stranded helix. To create the double helix, the nucleotides on the opposing strands of DNA form hydrogen bonds, where A always pairs with T, and C always pairs with G. The order of nucleotides makes up the genetic code and provides the instructions to make every protein within an organism.
Before proteins can be translated, the DNA is converted to RNA in a process called transcription. RNA uses the same nucleotide bases as DNA, except thymine is replaced by uracil (U). Not all regions of DNA code for proteins. In fact, most DNA is non-coding. These regions are transcribed into non-coding RNA (ncRNA) and play important roles in cellular processes and gene regulation. Regions that code for proteins are transcribed and processed into mature messenger RNA (mRNA), which is then translated into proteins.
Proteins are made up of amino acids. Each amino acid is encoded for by three nucleotides, termed a codon. There are 64 codon combinations and only 20 natural amino acids, so multiple codons can encode the same amino acid — a phenomenon called degeneracy. Refer to the amino acid table below for codon-amino acid pairs. Over the past two decades, researchers have also expanded the genetic code by redirecting some codons to encode for synthetic amino acids.
Plasmids and Recombinant DNA Technology
Chemistry techniques enable the isolation and purification of cellular components, such as DNA, but in practice this isolation is only feasible for relatively short DNA molecules. To isolate a particular gene from human chromosomal DNA, it would be necessary to isolate a sequence of a few hundred or a few thousand base pairs from the entire human genome. Digesting the human genome with restriction enzymes would yield about two million DNA fragments, which is far too many to isolate one specific DNA sequence. The field of recombinant DNA technology has overcome this obstacle by enabling the preparation of more manageable DNA fragments.
In 1952, Joshua Lederberg coined the term plasmid, in reference to any extrachromosomal heritable determinant. Plasmids are fragments of double-stranded DNA that typically carry genetic information and can replicate independently from chromosomal DNA. Although they can be found in archaea and eukaryotes, they play the most significant biological role in bacteria. Plasmids usually provide a benefit to the host, such as antibiotic resistance, and can be passed from one bacterium to another by a type of horizontal gene transfer called conjugation. Like chromosomal DNA, plasmid DNA is replicated upon cell division, and each daughter cell receives at least one copy of the plasmid.
By the 1970s, the combined discoveries of restriction enzymes, DNA ligases, and gel electrophoresis enabled the movement of specific fragments of DNA from one context to another, such as from a chromosome to a plasmid or vector DNA backbone. These tools are essential to the field of recombinant DNA, in which many identical DNA fragments can be generated. The combination of a DNA fragment with a plasmid or vector DNA backbone generates a recombinant DNA molecule, which can be used to study DNA fragments of interest, such as genes.
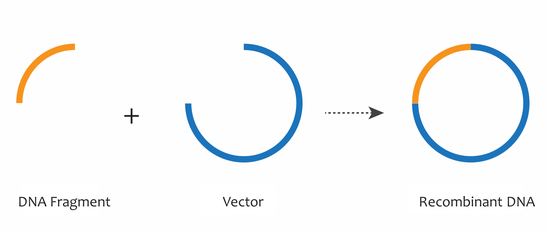
Working with Plasmids
Today, scientists can easily study and manipulate genes and other genetic elements using specifically engineered plasmids. These plasmids have become possibly the most ubiquitous tools in the molecular biologist’s toolbox because they:
- Are easy to work with — Plasmids are a convenient size (generally 1,000–20,000 base pairs) for physical isolation (purification) and manipulation. With current cloning technology, it is easy to create and modify plasmids containing the genetic element that you are interested in.
- Self-replicate — Once you have constructed a plasmid, you can easily make an endless number of copies of the plasmid using bacteria, which take up plasmids and amplify them during cell division. Because bacteria are easy to grow, divide relatively quickly, and exhibit exponential growth rates, plasmids can be replicated easily and efficiently in a laboratory setting.
- Are stable — Plasmids are stable long-term, either as purified DNA or within bacterial cells that have been preserved as glycerol stocks.
- Function in many species — Plasmids can drive gene expression in a wide variety of organisms, including plants, worms, mice, and cultured human cells.
- Have diverse applications — Although plasmids were originally used to understand protein coding gene function, they are now used for a variety of studies used to investigate promoters, small RNAs, and other genetic elements.
Plasmid Elements
Plasmids come in many sizes and vary broadly in their functionality. In their simplest form, plasmids require a bacterial origin of replication (ori), an antibiotic resistance gene, and at least one unique restriction enzyme recognition site (Figure 2). These elements allow for the propagation of the plasmid within bacteria, while allowing for selection against any bacteria not carrying the plasmid. Additionally, the restriction enzyme site(s) allow for the cloning of a DNA fragment of interest into the plasmid. Some common plasmid elements are summarized in the table below.
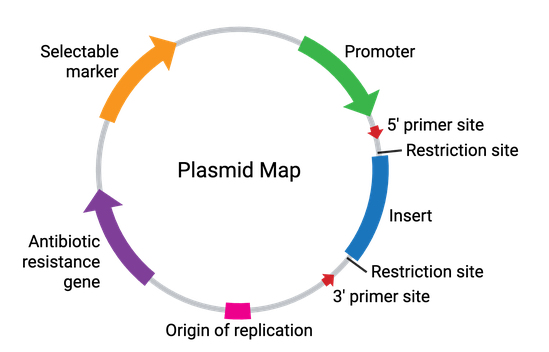
| Plasmid Element | Description |
|---|---|
| Origin of Replication (ori) | DNA sequence which directs the initiation of plasmid replication in bacteria by recruiting DNA replication machinery. The ori is critical for the ability of the plasmid to be copied (amplified) by bacteria. |
| Antibiotic Resistance Gene | Allows for selection of plasmid-containing bacteria by providing a survival advantage to the bacterial host. |
| Multiple Cloning Site (MCS) | A short segment of DNA which contains several restriction enzyme sites, enabling easy insertion of DNA by restriction enzyme digestion and ligation. As a general rule, the restriction sites in the MCS are unique and not located elsewhere in the plasmid backbone. In expression plasmids, the MCS is often located downstream from a promoter to drive expression of the inserted gene. |
| Insert | The gene, promoter, or other DNA fragment cloned into the MCS. The insert is typically the genetic element being studied using a particular plasmid. |
| Promoter Region | Drives transcription of the insert. The promoter recruits transcriptional machinery and can be specific to a certain species, tissue, or cell type. The strength of the promoter can control the level of insert expression, as a strong promoter directs high expression, whereas weaker promoters can direct low/endogenous expression levels. For more information about promoters, check out our promoters reference page. |
| Selectable Marker | Used to select for cells that have successfully taken up the plasmid and can be used to study the insert. This is different than selecting for bacterial cells that have taken up the plasmid. The selectable marker is typically in the form of another antibiotic resistance gene under the control of a non-bacterial promoter or a fluorescent protein to select or sort the cells by visualization or flow cytometry. |
| Primer Binding Site | A short, single-stranded DNA sequence used as an initiation point for PCR amplification or DNA sequencing of the plasmid. Primers can be utilized to verify the sequence of the insert or other regions of the plasmid. For a list of commonly used primers, check out our sequencing primers page. |
Types of Plasmids
Plasmids are versatile and can be used in many different ways by scientists. The combination of elements often determines the type of plasmid and dictates how it might be used in the lab. Regardless of type, plasmids are generally propagated, selected for, and their integrity verified prior to use in an experiment.
| Plasmid Type | Description | Addgene Resource(s) |
|---|---|---|
| Cloning Plasmids | Used to facilitate the cloning of DNA fragments. Cloning vectors tend to be very simple, often containing only a bacterial resistance gene, origin of replication, and an MCS. They are small and optimized to help in the initial cloning of a DNA fragment. Commonly used cloning vectors include Gateway entry vectors and TOPO cloning vectors. | Empty Backbones, Cloning Kits |
| Expression Plasmids | Used for gene expression, usually for the purposes of gene study. Expression vectors must contain a promoter sequence, a transcription terminator sequence, and the inserted gene. An expression vector can also include an enhancer sequence which increases the amount of protein or RNA produced. Expression vectors can drive expression in various cell types (mammalian, yeast, bacterial, etc.), depending largely on which promoter is used to initiate transcription. | Empty Backbones |
| Gene Knock-down Plasmids | Used for reducing the expression of an endogenous gene. This is frequently accomplished through expression of an shRNA targeting the mRNA of the gene of interest. These plasmids have promoters that can drive expression of short RNAs. | Mammalian shRNA Tools |
| Genome Engineering Plasmids | Used to target and edit genomes. Genome editing is most commonly accomplished using CRISPR technology. CRISPR is composed of a DNA endonuclease and guide RNAs that target specific locations in the genome. For more information on CRISPR check out our CRISPR Guide. | CRISPR Plasmids, Cre-lox and Other Recombinases, TALEN Plasmids and Kits, Genome Engineering Kits |
| Reporter Plasmids | Used for studying the function of genetic elements. These plasmids contain a reporter gene (e.g., luciferase or GFP) that offers a readout of the activity of the genetic element. For instance, a promoter of interest could be inserted upstream of the luciferase gene to determine the level of transcription driven by that promoter. | Fluorescent Protein Plasmids, Luciferase Plasmids |
| Viral Plasmids | These plasmids are modified viral genomes that efficiently deliver genetic material into target cells. You can use these plasmids to create viral particles (most commonly lentiviral, retroviral, AAV, or adenoviral particles) that can infect your target cells at a high efficiency. | Viral Vectors, Viral Services |
Molecular Cloning
Many common plasmids used in the field of recombinant DNA technology have been optimized for studying and manipulating genes. For instance, most plasmids are replicated in E. coli and are relatively small (∼3,000–6,000 base pairs) to enable easy manipulation. Typically, plasmids contain the minimum essential DNA sequences (or plasmid elements) necessary for their intended purpose.
When a plasmid exists extrachromosomally in E. coli, it is replicated independently and segregated to the resulting daughter cells. These daughter cells are called clones, since they contain the same genetic information as the parental cell. The plasmid DNA is similarly referred to as cloned DNA, and this process of generating multiple identical copies of a recombinant DNA molecule is known as DNA or molecular cloning. The advent of molecular cloning enabled scientists to break chromosomes down to study their genes, marking the birth of molecular genetics.
To learn more about different types of cloning methods, check out our guide on molecular cloning techniques.
Common Bacteria Strains for Propagating Plasmids
E. coli are gram-negative, rod-shaped bacteria naturally found in the intestinal tract of animals. There are many different naturally occurring strains of E. coli, some of which are deadly to humans. Most of all common, commercial lab strains of E. coli used today are descended from two individual isolates, the K-12 strain and the B strain. K-12 has led to the common lab strains MG1655 and its derivatives DH5alpha and DH10b (also known as TOP10) among others, while the B strain gave rise to BL21 and its derivatives.
We have included a small number of E. coli strains below and recommend checking out Addgene’s blog posts about common E. coli lab strains and E. coli strains specialized for protein expression for additional strain-related information and a more extensive strain list.
| Strain | Vendor(s) | Genotype |
|---|---|---|
| BL21 | Invitrogen; New England BioLabs | E. coli B F dcm ompT hsdS(rB mB) gal |
| ccdB Survival | Invitrogen | F- mcrA Δ(mrr-hsdRMS-mcrBC) Phi80lacZΔM15 Δ-lacX74 recA1 araΔ139 D(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG tonA::Ptrc ccdA |
| DB3.1 | Invitrogen | F- gyrA462 endA Δ(sr1-recA) mcrB mrr hsdS20 (rB- mB-) supE44 ara14 galK2 lacY1 proA2 rpsL20(StrR) xyl5 lambda- leu mtl1 |
| DH5alpha | Invitrogen | F- Phi80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17(rk-, mk+) phoA supE44 thi-1 gyrA96 relA1 tonA |
| EPI300 | LGC Biosearch Technologies; MilliporeSigma | F- mcrA Δ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL (StrR) nupG trfA dhfr |
| JM109 | Addgene; Promega | e14-(McrA-) recA1 endA1 gyrA96 thi-1 hsdR17(rK- mK+) supE44 relA1 Δ(lac- proAB) [F traΔ36 proAB lacIqZΔM15] |
| NEB Stable | New England Biolabs | F' proA+B+ lacIq ∆(lacZ)M15 zzf::Tn10 (TetR) ∆(ara-leu) 7697 araD139 fhuA ∆lacX74 galK16 galE15 e14- Φ80dlacZ∆M15 recA1 relA1 endA1 nupG rpsL (StrR) rph spoT1 ∆(mrr-hsdRMS-mcrBC) |
| Stbl3 | Invitrogen | F- mcrB mrr hsdS20 (rB-, mB-) recA13 supE44 ara-14 galK2 lacY1 proA2 rpsL20 (StrR ) xyl-5 λ- leu mtl-1 |
| Top10 | Invitrogen | F- mcrA Δ(mrr-hsdRMS-mcrBC) Phi80lacZM15 Δ-lacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG |
Common Antibiotics for Plasmid Selection
Each bacterium can contain multiple copies of an individual plasmid, and ideally would replicate these plasmids upon cell division in addition to their own genomic DNA. Because of this additional replication burden, the rate of bacterial cell division is reduced, as it takes more time to copy this extra DNA. Because of this reduced fitness, bacteria without plasmids can replicate faster and out-populate bacteria with plasmids, thus selecting against the propagation of these plasmids through cell division.
To ensure the retention of plasmid DNA in bacterial populations, plasmids are designed to include an antibiotic resistance gene, which when expressed, allows only plasmid-containing bacteria to grow in or on media containing that antibiotic. These antibiotic resistance genes not only give the scientist an easy way to detect plasmid-containing bacteria, but also provide those bacteria with selective pressure to maintain and replicate your plasmid over multiple generations. It is important to distinguish that the antibiotic resistance gene is under the control of a bacterial promoter, and is thus expressed only in bacteria by bacterial transcriptional machinery. For more information on antibiotic resistance and additional antibiotics, see our blog post on antibiotic resistance genes.
Below you will find a few antibiotics commonly used in the lab and their recommended concentrations. We suggest checking your plasmid's datasheet or the plasmid map to confirm which antibiotic(s) to add to your LB media or LB agar plates. For more information on growing bacterial cultures with antibiotics, see Addgene’s protocol on inoculating bacterial cultures.
| Antibiotic | Recommended Stock Concentration | Recommended Working Concentration |
|---|---|---|
| Ampicillin | 100 mg/mL | 100 µg/mL |
| Carbenicillin* | 100 mg/mL | 100 µg/mL |
| Chloramphenicol | 25 mg/mL (dissolve in Ethanol) |
25 µg/mL |
| Hygromycin B | 200 mg/mL | 200 µg/mL |
| Kanamycin | 50 mg/mL | 50 µg/mL |
| Spectinomycin | 50 mg/mL | 50 µg/mL |
| Tetracycline | 10 mg/mL | 10 µg/mL |
*Carbenicillin can be used in place of ampicillin.
DNA Sequencing for Plasmid Verification
Scientists often sequence DNA to identify the order of nucleotide bases in a particular DNA strand. Sequencing DNA and understanding the genetic code allows scientists to study gene function as well as identify changes or mutations that may cause certain diseases. Sequencing DNA is extremely important when verifying plasmids to ensure each plasmid contains the essential elements to function and the correct gene of interest.
Sequencing technologies like Sanger sequencing and next-generation sequencing (NGS) take advantage of the process of DNA replication in vitro. During replication, the DNA helix is unwound by a helicase, the enzyme that breaks the hydrogen bonds between nucleotides on opposing strands. A DNA polymerase then binds to the single strand of DNA. Using this as a template, the polymerase migrates down the DNA, adding nucleotides according to the sequence of the complementary strand. Mimicking the DNA replication process requires the four nucleotides (dNTPs), a DNA polymerase enzyme, the template DNA to be copied, and a primer. A primer is a small piece of DNA, approximately 18–22 nucleotides, that binds to complementary DNA and acts as a starting point for the DNA polymerase. Thus to replicate a piece of DNA in vitro, you have to know some of its sequence to design a effective primer.
To learn more about the sequencing technologies Addgene uses to maintain the quality standards of our plasmid collection, see our blog post on QC sequencing technologies at Addgene.
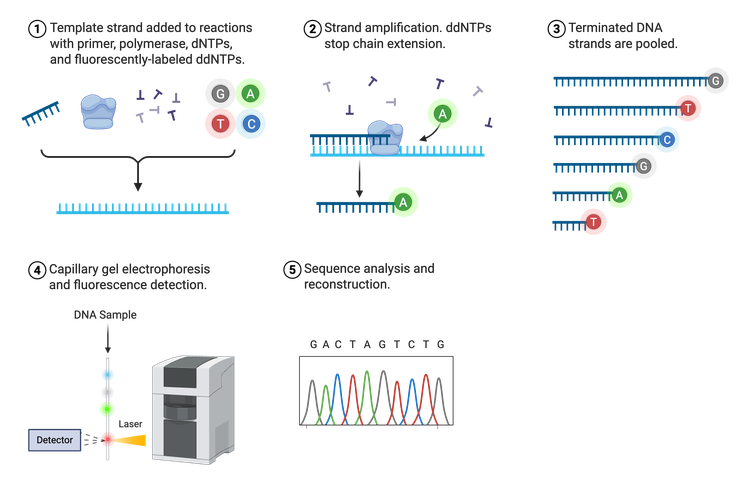
Sanger Sequencing
In 1975, Frederick Sanger developed the process termed Sanger sequencing, sometimes referred to as chain-termination sequencing or dideoxy sequencing (Figure 3). Sanger sequencing relies on the random incorporation of modified, fluorescently-tagged bases during in vitro DNA replication in addition to the normal A, T, C, or G nucleotides. The four standard bases (dNTPs) are tagged with a different fluorophore so they can be distinguished from one another. These tagged dNTPs also lack a binding site for the next nucleotide (denoted as ddNTPs), halting synthesis after their incorporation.
Sanger sequencing requires a lot of DNA because the ultimate goal is to have a fluorescently-tagged nucleotide at each position in the DNA sequence. Thus, the final result is a group of newly synthesized DNA strands of varying lengths whose last nucleotide is labeled. These DNA strands are separated by size, read using fluorescence detection, and the sequence outputted in the form of a chromatogram. Sanger can sequence approximately 500–1,000 bases downstream of the known primer region with very few errors, making it an efficient and reliable sequencing method.
Next-generation Sequencing
Although Sanger sequencing is quick and efficient, it is low-throughput and can only sequence short pieces of DNA. This makes it difficult to sequence an entire plasmid or genome. One Sanger sequencing reaction would give you only 20 pieces of a 2,000 piece puzzle. A scientist would need to run potentially hundreds of Sanger sequencing reactions on different pieces of DNA to be able to assemble the whole puzzle. That’s where NGS (sometimes referred to as “second-generation sequencing”) comes in. NGS is a high-throughput, multi-parallel sequencing platform that can generate sequencing data for up to 600 billion bases in one reaction. In other words, NGS can give you most of the puzzle pieces in only a few reactions. There are multiple approaches to acquire NGS, but one of the most common is the Illumina NGS platform for short-read sequencing. This is the platform Addgene primarily uses for sequencing plasmids deposited at Addgene.
The actual process of Illumina NGS is not that different from Sanger sequencing (Figure 4). Like Sanger, NGS utilizes modified, fluorescently-tagged nucleotides. During Illumina NGS, a long piece of DNA is fragmented into small pieces, labeled with a short DNA barcode, and amplified. These DNA fragments are attached to a glass slide so that different fragments of DNA, or templates, are spatially separated. These attached DNA templates are then amplified again, producing ~1,000 copies of each template. Each template is then replicated using the modified bases and a microscope captures the fluorescent color that is emitted each time a base is added. Again, each base (A,C,T, or G) is labeled with a different color, making it easy to identify the order of the DNA strand. Unlike Sanger, however, these modified bases can be converted back to a regular base and thus do not halt the reaction. Illumina NGS, therefore does not require any “normal” (nonlabeled) bases in the reaction. All the sequenced templates are then aligned to each other to assemble the entire sequence using bioinformatic methods. It is important to note that NGS platforms in general do not require a specific primer for your DNA of interest, so even a completely unknown piece of DNA can be sequenced.
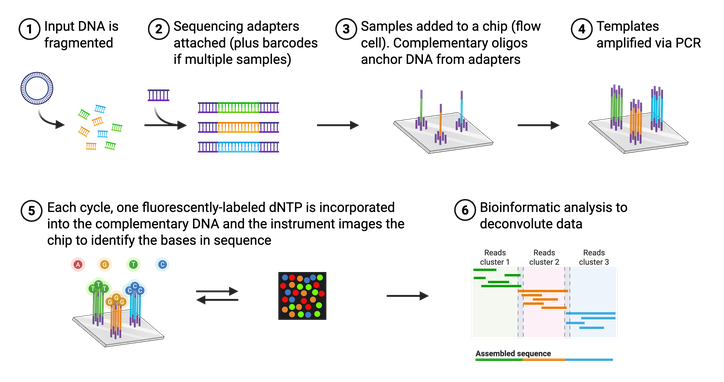
At Addgene, all incoming plasmids are sequenced with NGS during our quality control process. NGS allows us to sequence entire plasmids, providing scientists with even more information to aid in the reproducibility of scientific research.
Long-read Sequencing
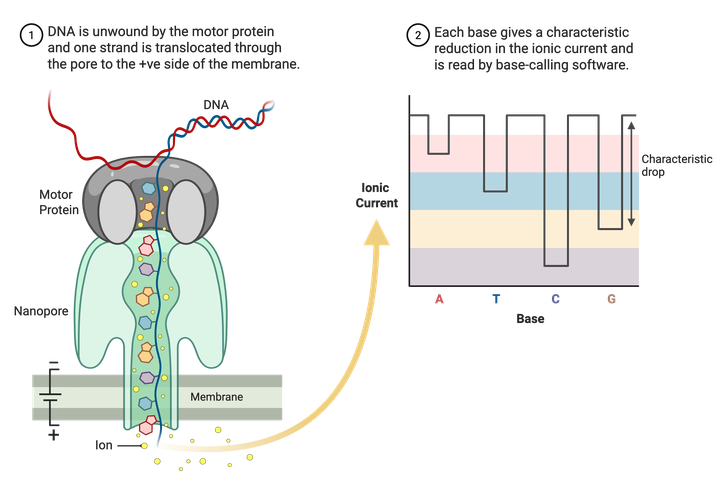
Sanger sequencing and Illumina NGS are limited to shorter reads of DNA, usually a couple hundred base pairs at most. Long-read sequencing technologies, like Nanopore, can read thousands to potentially millions of base pairs at a time. Nanopore uses specialized membranes containing nanopores (Figure 5). A motor protein then unwinds double-stranded DNA, and a single stand is pulled through the pores. An electrical current runs through the porous membrane, and the changes in this current are recorded as the DNA is pulled through. The changes in current are correlated with specific nucleotides and allow for the assembly of a complete sequence. Using this method, whole plasmid sequences can be read in one run.
Resources
Genetic Code
The genetic code can be defined as a set of rules for translating the information encoded by DNA and RNA into proteins. DNA is comprised of four nucleotides: Adenine (A), Thymine (T), Cytosine (C) and Guanine (G). In the double helix, A always pairs with T and C always pairs with G. RNA, on the other hand, consists of Adenine, Cytosine, Guanine and Uracil (U). Uracil replaces thymine in RNA molecules. Every 3 nucleotides (codons) in a DNA sequence encodes for an amino acid. The genetic code is degenerate thus multiple codons code for each amino acid. There are 20 amino acids plus a start and stop codon.
Below you will find helpful resource tables about the genetic code. This table includes the nucleotide and amino acid code in addition to ambiguous bases and common epitope tags. Ambiguous bases are included in a DNA sequence when sequencing is not 100% efficient and the machine cannot distinguish between the four labeled nucleotides, or in biologically relevant contexts like when a DNA-binding protein has some flexibility in the recognition sequence. Epitope tags, on the other hand, are commonly used in molecular cloning to tag a gene within a plasmid.
DNA and RNA
| Single Letter Code: Primary bases | Nucleobase |
|---|---|
| A | Adenine |
| C | Cytosine |
| G | Guanine |
| T | Thymine |
| U | Uracil |
| Single Letter Code: Ambiguous bases | Nucleobase |
|---|---|
| B | C, G, or T |
| D | A, G, or T |
| H | A, C, or T |
| K | G or T |
| M | A or C |
| N | A, T, C, or G |
| R | A or G |
| S | C or G |
| V | A, C, or G |
| W | A or T |
| Y | C or T |
Amino Acids
| Name | Three Letter Code | Single Letter Code | Codons (RNA) |
|---|---|---|---|
| Alanine | Ala | A | GCU, GCC, GCA, GCG |
| Arginine | Arg | R | CGU, CGC, CGA, CGG, AGA, AGG |
| Asparagine | Asn | N | AAU, AAC |
| Aspartic Acid | Asp | D | GAU, GAC |
| Cysteine | Cys | C | UGU, UGC |
| Glutamine | Gln | Q | CAA, CAG |
| Glutamic Acid | Glu | E | GAA, GAG |
| Glycine | Gly | G | GGU, GGC, GGA, GGG |
| Histidine | His | H | CAU, CAC |
| Isoleucine | Ile | I | AUU, AUC, AUA |
| Leucine | Leu | L | UUA, UUG, CUU, CUC, CUA, CUG |
| Lysine | Lys | K | AAA, AAG |
| Methionine | Met | M | AUG |
| Phenylalanine | Phe | F | UUU, UUC |
| Proline | Pro | P | CCU, CCC, CCA, CCG |
| Serine | Ser | S | UCU, UCC, UCA, UCG, AGU,AGC |
| Threonine | Thr | T | ACU, ACC, ACA, ACG |
| Tryptophan | Trp | W | UGG |
| Tyrosine | Tyr | Y | UAU, UAC |
| Valine | Val | V | GUU, GUC, GUA, GUG |
| Start | AUG* | ||
| Stop | UAG (amber), UGA (opal), UAA (ochre) |
*AUG is the most common start codon. Alternative start codons include CUG in eukaryotes and GUG in prokaryotes.
Additional Resources
Content last reviewed: 22 October 2025