
Antibody Guide
Affinity reagents are molecular tools used in the lab to specifically and tightly bind to other proteins. Antibodies, produced as part of the immune system’s response to foreign bodies, are the most commonly used affinity reagents.
Antibodies are Y-shaped proteins, naturally produced by the immune system to recognize and bind foreign bodies. Antibodies can be used in the lab to quantify, identify, separate, and locate proteins of interest. The protein that an antibody recognizes is called an antigen, while the specific binding site is referred to as an epitope.
Production
Antibodies are typically produced in one of three different methods: in animals, in cultured immune cells, or through the use of recombinant plasmids.
- Animals - Antibodies produced in animals make use of the animal’s immune system. The antigen of interest is introduced to the animal, which then mounts an immune response to it. Once the immune response has developed, the antibodies are collected, tested against the antigen, packaged, and sold. This is the most cost-efficient way to produce antibodies, with the highest variability in antibodies produced.
- Cells - Antibodies can also be produced in cell cultures using hybridomas, cell cultures created by fusing B cells with myeloma cells (immortalized B cells). The genetic reorganization that creates variety in the antibody allowing for their extreme specificity occurs in the B cell itself prior to antibody production; therefore, each B cell only produces one type of antibody. Monocultures of B cells isolated from animals immunized as described above are used to create hybridomas, which produce large amounts of a single antibody with little variation.
- Recombinant plasmids - Antibodies can be produced from plasmids, with an antibody’s genetic code inserted into the plasmid. This produces the least amount of variation between antibodies, allows for the antibody’s identity to be easily confirmed via sequencing, and enables the variable region and constant regions of the antibody to be independently manipulated. Antibodies produced this way are called recombinant antibodies. Recombinant antibodies are created by cloning antibody genes from specific hybridoma cells into plasmids. Plasmids are then transfected into cells and induced to express antibodies. Antibodies are released into the growth medium and can either be used while suspended in the original media or isolated and placed in a buffer.
Antibody Structure
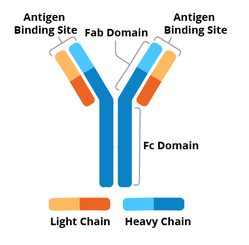
A standard antibody is made up of four protein chains: two large heavy chains and two small light chains. These four domains come together to form a “Y” shape, as shown in Figure 1. The two arms of the Y structure are responsible for binding the antigen, and are called the antigen binding fragments (Fab). The tips of the Fab fragments contain highly variable regions, shown in light blue and light orange, that bind to a specific antigen, triggering an immune response. The stem of the Y structure and the lower part of the arms is called the constant region, and is shown in dark blue and dark orange.
The constant region is a stable part of the antibody, which determines the isotype of the antibody. B cells, the immune cell responsible for producing antibodies, can change the constant region, and therefore change the isotype of antibodies they produce. Just as antibodies are capable of recognizing and binding to epitopes on antigens, they are also capable of recognizing and binding epitopes on constant regions of other antibodies. This has useful research applications, and is covered in the following Isotypes section.
The variable region of the antibody is the antigen binding site. In B cells, the genetic region responsible for the variable region goes through a randomized reorganization process, creating a unique binding site. This process, known as V(D)J recombination, allows for a wide variety of antibodies to be produced in a single individual, giving a high probability that an antibody will be able to recognize and bind to any given antigen. A mature B cell can clone itself to enable higher production of a single antibody, but each B cell can only produce one variable region.
Isotypes
The mammalian immune system produces five classes of antibodies, known as isotypes. An isotype is determined by the antibody’s constant region - specifically, it is determined by a region called the “heavy chain constant region” or Fc. Different isotypes play different roles in the immune response, and can be used to increase flexibility or specificity in antibody-based methods.
Antibodies can recognize and bind other antibodies using epitopes present on the Fc region. This allows for the amplification of signals by having multiple secondary antibodies binding to a primary antibody which is bound to your protein of interest. Secondary antibodies can be isotype-specific or even isotype subclass-specific. This specificity can be used to ensure secondary antibodies bind to a specific primary antibody in assays with multiple targets, and is described in more detail in the Multiplex Assays section below. The following table describes the five human isotypes. Some isotypes naturally form multi-antibody units, while others exist as monomers. Each antibody class therefore has a valency, or number of binding sites available per antibody unit, which is typically the number of antibodies multiplied by two, as each antibody has two binding sites.
| Isotype | Description | Configuration and Valency | Subclasses |
|---|---|---|---|
| IgA | Prevalent antibody in secretions (e.g. tears, saliva, mucus) where it protects mucosal membranes | Dimer with a valency of 4 | IgA1; IgA2 |
| IgD | Coexpressed with IgM on the surface of most immature B cells; function is unknown | Monomer with a valency of 2 | |
| IgE | Protects against parasites and is responsible for driving allergic reactions such as anaphylactic shock | Monomer with a valency of 2 | |
| IgG | The most abundant isotype in blood, also found in tissues; predominant isotype in secondary immune response | Monomer with a valency of 2 | IgG1; IgG2; IgG3; IgG4 |
| IgM | Produced during the primary immune response; found mostly in blood | Pentamer with a valency of 10 |
Antigen Specificity
Antibodies are available in two types of antigen specificity: monoclonal, meaning many copies of one clone that are specific to a certain epitope, or polyclonal, meaning there are many clones that are specific to multiple epitopes on one antigen.
Monoclonal antibodies are generated from cultured B cells, where each culture should produce the same antibody clone, or from plasmid-based expression. Monoclonal antibodies are necessary in cases where extreme specificity is important. Their epitope specificity allows binding to a specific configuration of a protein or a protein:protein complex. Due to their increased manufacturing requirements, monoclonal antibodies are typically more expensive than polyclonal antibodies.
Environmental factors, binding partners, location in the cell, and sample processing can hide or reveal epitopes on an antigen. Monoclonal antibodies only bind a single epitope, so it is important to choose a monoclonal antibody that will recognize an available epitope on your protein of interest.
Polyclonal antibodies are typically collected directly from animals and contain antibodies targeting various epitopes on the same antigen. Polyclonal antibodies are often more environmentally stable and more cost-effective. They are useful when epitope specificity is not a concern or in cases where you may want to identify or collect the protein in any form it may be present in. However, polyclonals can vary significantly from lot to lot, as immune responses have high inter- and intra-individual variability.
Other Affinity Reagents
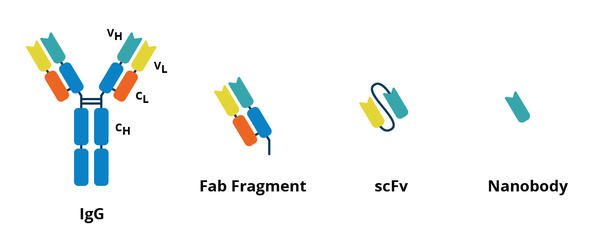
There are several alternatives to antibodies, ranging from antibody fragments (shown in Figure 2) to completely unrelated molecules. Examples include:
- scFv - Single-chain fragment variables (scFvs) contain only the variable heavy chain and variable light chain regions of an antibody. They are small enough to be produced in bacteria (~25 kD), but tend to have lower affinity and longevity.
- Single-domain antibodies - Single-domain antibodies (sdAbs), also called nanobodies, are ~12-15 kD single heavy domain fragments derived from a camelid antibody called a heavy chain antibody (HCAb). Their small size allows them to bind to epitopes that are not accessible to antibodies. Detection of sdAbs is usually achieved by conjugation of a signaling molecule directly to the sdAb, or by using a secondary against a tag, such as His, incorporated in the sdAb. sdAbs can have weak signals due to their monovalent nature.
- Darpins - Darpins are genetically engineered small proteins derived from ankyrin repeat proteins. These are antibody mimics instead of modified antibodies and are not closely related to antibodies.
- Monobodies - Monobodies are synthetic binding proteins which use the Fibronectin type III domain as their scaffolding instead of the antibody constant region.
- Minibodies - Minibodies are engineered antibody fragments consisting of two scFvs bound to a CH3 domain.
- Fab fragments - Fab fragments are created by enzymatically cleaving the variable regions of an antibody (each Fab fragment is a single arm of the Y). They can be conjugated to signaling molecules and are often used in clinical applications.
- Diabodies - Diabodies contain two Fab fragments, each recognizing a different epitope, bound by short peptide linkers. Diabodies can be used for assembling protein nanostructures.
Antibody Handling
Storage
Check the manufacturer’s instructions for exact storage requirements. As a guideline, most antibodies can be stored at 4 °C for short-term storage. For long-term storage they should be stored at -20 °C in aliquots of at least 10 µL. Antibodies cannot tolerate freeze-thaw cycles.
Here are a few tips for storing your antibody:
- Antibodies conjugated to fluorophores should never be stored below 4 °C.
- Certain isotype subclasses, such as IgG3, aggregate after thawing and therefore should be kept at 4 °C.
- Record and track lot numbers for all antibodies used in the lab.
- Check for and record any expiration dates.
Buffers
Buffers are a concern for conjugating and signaling molecules, as some buffer components can interfere with the conjugate or conjugation process.
Sodium azide, an antimicrobial agent, can block some amines needed for conjugation reactions and should be removed from the buffer before the reaction. However, it can be added back to buffers after the conjugation reaction is complete. HRP-conjugated antibodies should not be stored in sodium azide, as it acts as an inhibitor to HRP. In those cases, thimerosal can be used as an antimicrobial agent.
Other chemical interferences with conjugating reactions are possible. It is important to research your reaction and buffer before conjugating your antibodies.
Antibody Visualization
The ability of an antibody to specifically bind to a protein is powerful, but in order to utilize it in the lab, researchers must be able to detect if and where an antibody has bound. Signaling molecules, proteins that emit a measurable light or color in response to a specific stimulus, can be conjugated to antibodies in order to allow detection of the antibody:protein complex. The most commonly used types of signaling molecules are horseradish peroxidase (HRP) and fluorophores.
Horseradish peroxidase (HRP)
HRP performs a chemiluminescent reaction in the presence of several different substrates. The luminescence produced by the reaction can be detected either digitally by an imaging machine or by X-ray film. HRP cannot be used to distinguish between different antibodies in the same reaction. It is therefore often used in western blots, which use size to differentiate between proteins.
HRP is also used in ELISAs, which only measure a single antibody signal. Here, HRP is used as a colorimetric, in which the reaction produces a colored product that accumulates over time which is relative to the amount of enzyme present in each well. In colorimetric assays, protein amount can be determined by correlating the amount of color in a well to a standard curve with known amounts of protein.
Fluorophores
Fluorophores are light-sensitive molecules which emit a specific wavelength of light after excitation by light of a specific wavelength. While more expensive than HRP, fluorophores are available in a range of colors activated by different wavelengths, allowing for easy differentiation between multiple antibodies. They are often used in assays that have multiple proteins to detect simultaneously.
Conjugation
The process of attaching signaling molecules to an antibody is done through a conjugation reaction. Many antibodies can be purchased already conjugated, which can save significant time in the lab. However, if the antibody you want is not available pre-conjugated, or is not available conjugated to your signaling molecule of choice, you can conjugate antibodies in the lab. The complexity of this can vary significantly based on the antibody and conjugate chosen.
Signal Amplification
The amount of signal needed from a specific antibody can vary depending on the assay. The ability of secondary antibodies to recognize and bind to the Fc region of primary antibodies allows for signal amplification through the indirect detection method.
In the direct detection method, the signaling molecule is conjugated to the primary antibody, which recognizes and binds to the protein of interest. Signal strength is determined by how many antibodies bind the protein of interest, while the number of proteins that you can detect in a single assay is limited by the number of signaling molecules that can be distinguished in your application.
The indirect detection method uses an unconjugated primary antibody and a conjugated secondary antibody specific to your primary antibody. Multiple secondary antibodies will bind to a single primary antibody, which significantly increases the signaling molecule to epitope ratio. Secondary antibodies can distinguish between different species of origin, isotypes, and isotype subclasses of a primary antibody, which becomes the limiting factor for the number of proteins you can detect in a single assay.
Overview of Antibody Applications
Antibody-based applications can be generally classed into four categories:
-
Quantification - used to quantify relative amounts of protein. Includes:
- Western blot
- ELISA
-
Capture - used to isolate specific proteins or protein complexes. Includes:
- Immunoprecipitation (IP)
- Chromatin immunoprecipitation (ChIP)
-
Visualization - used to visualize protein location. Includes:
- Immunofluorescence (IF)
- Immunohistochemistry (IHC)
- Immunocytochemistry (ICC)
-
Cell Sorting - used to identify, analyze, capture, and sort single cells. Includes:
- Flow cytometry
Read on to learn more about the applications common to each category, including an outline of the basic steps needed for each application, important controls, and any special considerations to think about. For step-by-step instructions of many of these applications, visit the Antibody section of the Addgene Protocols page.
Antibody Applications - Quantification Methods
Quantification methods, such as western blots or ELISAs, are used to quantify relative amounts of protein.
Western blot
Western blots use antibodies to identify denatured proteins separated by size on an SDS-PAGE gel. Though relative quantification can be done, western blots primarily show a protein’s presence or absence.
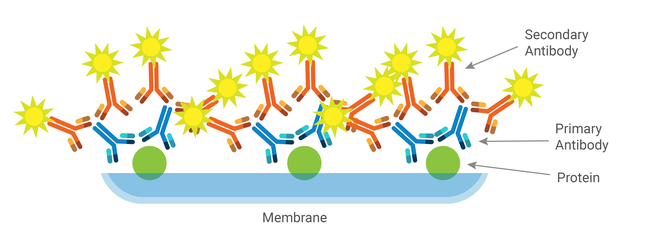
Overview of application
- Collect and lyse samples to make proteins available. Tissue samples may need additional processing steps, such as sonication.
- Denature proteins, using heat and/or chemicals, and run on an SDS-PAGE gel to separate proteins by size.
- Transfer proteins from the gel to a membrane.
- Incubate the membrane with an unconjugated primary antibody.
- Wash and block the membrane to remove excess antibody and prevent unwanted binding.
- Incubate the membrane with a secondary antibody conjugated with HRP.
- Wash and block the membrane.
- Activate the HRP with a substrate kit.
- Image the membrane using either X-ray film in a darkroom or an image capture machine.
Special considerations
The primary antibody must be able to recognize the denatured configuration of the protein. For western blots, monoclonal antibodies tend to have less cross-reactivity and give lower background signals than polyclonal antibodies. However, polyclonal antibodies are more sensitive to lower amounts of protein in the sample.
Western blots should be run with your protein of interest and at least one ubiquitously expressed protein such as GAPDH or tubulin (sometimes referred to as housekeeping genes). This allows for relative comparison of expression between different samples, by normalizing protein expression to the controls before comparison. Alternatively, the samples can be normalized to a total protein measurement. Both methods will also serve as a control for protein expression and assay efficacy.
ELISA
Enzyme-linked immunosorbent assay (ELISA) is a plate-based assay that uses standard curves to quantify exact amounts of proteins. ELISAs quantify a single protein per assay and typically use HRP as a signaling molecule.

Overview of application
- Coat antigens to the well of a plate, either by charge interactions (direct) or by a capture antibody (sandwich).
- Block unused surface-binding sites in the wells with a blocking protein such as BSA, followed by several wash steps.
- Add primary antibody to the plate. After incubation, wash to remove unbound antibody. When the primary antibody is conjugated, it is often called the detection antibody.
- If using an indirect detection method, add a secondary antibody, followed by incubation and wash steps.
- Activate the conjugate and read the output via a plate reader.
Special considerations
The two most common types of ELISAs are direct or sandwich ELISAs. For direct ELISAs, an antigen or protein of interest is attached to the well of a 96-well plate. A conjugated primary antibody is used to bind to the antigen and the conjugate, typically HRP, is activated and its output measured. This method can be performed using the indirect detection method via a conjugated secondary antibody.
In a sandwich ELISA, a capture antibody, bound to a surface, is used to capture the protein of interest and immobilize it. A primary or detection antibody is added and binds to the bound protein. If using the indirect sandwich method, a conjugated secondary antibody is added. The conjugate is activated and the output is read.
A standard curve with known concentrations of protein should be used for each assay run. The standard curve should contain a range inclusive of all expected protein concentrates in the sample.
Controls for quantification methods
Quantification methods should be run with: a positive control, a sample previously verified to contain your protein of interest (in ELISAs, the standard curve can serve as the positive control); and a negative control, a sample known not to contain your protein of interest. The negative control should be biological (no protein expressed), which controls for your antibody’s specificity. A blank (contains water or buffer instead of a biological sample), which captures background expression and controls for cross-contamination, should also be included.
Antibody Applications - Capture Methods
Capture methods such as immunoprecipitation (IP) and chromatin immunoprecipitation (ChIP) allow you to isolate specific proteins or protein complexes for downstream analysis. They are powerful tools for understanding protein or protein interactions in a specific timeframe or context.
Immunoprecipitation (IP)
IP uses immobilized antibodies to isolate a specific protein out of a complex mix of proteins or cell materials. There are two primary methods used, using agarose or magnetic beads, but both rely on the same general strategy.
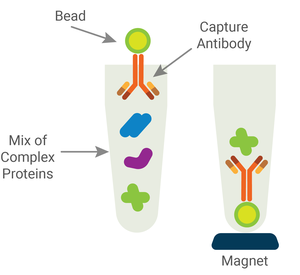
Overview of application
- Process the tissue or cell culture samples to extract proteins.
- Homogenize (tissues only), lyse, and then collect protein extract by centrifugation or affinity columns.
- Mix samples with a primary antibody, which is immobilized on agarose or magnetic beads.
- Using centrifugation (for agarose beads) or a magnet (for magnetic beads), collect the antibody:protein complexes.
- Wash to remove unbound antibodies and proteins.
- Elute the target protein.
Special considerations
The primary antibody must be able to recognize the protein in its native conformation.
Attachment of the antibody to the beads is typically done using Protein A, Protein G, or the recombinant Protein A/G, which are bacterial proteins that bind to the Fc portion of an antibody. However, they are not suitable for isolating protein from serums.
Chromatin immunoprecipitation (ChIP)
This variation of IP uses antibodies to capture protein:DNA complexes. After capture, the DNA is released from the complex and can be sequenced and amplified for analysis.
Overview of application
- Process tissue or cell culture samples to extract proteins.
- Homogenize (tissues only) and lyse.
- Cross-link the samples with formaldehyde or UV light.
-
Shear the samples using sonication to break DNA up into fragments of 300-1000 bps in length.
Note: This sonication process may need to be optimized for individual assays. - Clear cell debris by sedimentation.
- Proceed with IP using a primary antibody for your protein of interest, as described in the IP section above.
- Reverse the crosslink, isolate DNA, and perform downstream analysis.
Special considerations
One type of ChIP assay, called native ChIP, does not use cross-linking and instead relies on strong interactions between DNA and protein. Micrococcal nuclease digestion is used to shear the DNA in these assays. This method is suitable for DNA:histone modifier interactions, which have strong binding between the protein and DNA.
Some ChIP protocols use enzyme digestion instead of sonication. This approach is gentler but results in non-random fragmentation of the DNA.
Controls for capture methods
Capture methods should use a positive control and a negative control. The negative control may be a no-antibody control in which a sample is processed without antibodies, and/or one with a protein the antibody does not bind to.
In addition to the control used in standard IP, ChIP should additionally use a beads-only control which runs a water/buffer sample (with beads) to determine background signal levels. In ChIPseq, an input control will need to be collected before immunoprecipitation.
Antibody Applications - Visualization Methods
Visualization methods such as immunofluorescence (IF), immunohistochemistry (IHC), and immunocytochemistry (ICC) use labeled antibodies to visualize protein location in a cell or tissue sample.
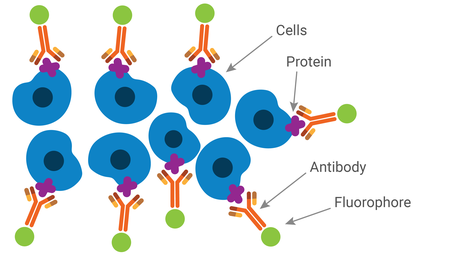
Immunofluorescence (IF)
This method uses fluorophore-conjugated antibodies to bind to and visualize proteins of interest in a cell or in tissue samples. Like most antibody methods, it can be done using either an indirect or direct method.
Since IF uses fluorophore color to differentiate between the targets of interest, if your fluorophore is bound to a secondary antibody, the secondary antibodies must be able to distinguish between the primary antibodies in multiplex assays. This is done by using different isotypes for each primary antibody and secondary antibodies specific to each isotype.
Overview of application
- Fix your samples.
- Prepare your samples using permeabilization, antigen retrieval, and/or sectioning.
- Block to reduce non-specific binding of antibodies.
- Add primary antibody, incubate, and wash.
- Add secondary antibody, incubate, and wash.
- Preserve sample and image using a microscope.
Special considerations
Many of the steps in this protocol vary depending on assay and microscopy techniques used, so be sure to read materials specific to your assay.
Most IF techniques use a nuclear stain, such as DAPI, to identify individual cells. These stains also emit fluorescent emissions and can be used in conjunction with antibody labeling. This means the majority of IF assays are considered multiplex assays, even if they only have one protein of interest.
Immunohistochemistry (IHC) and immunocytochemistry (ICC)
IF can refer to both immunohistochemistry and immunocytochemistry when fluorescent signaling molecules (also called dyes) are used. In IHC, tissue sections are assayed and the extracellular matrix remains intact. The signaling molecule may be a fluorophore or an enzyme. In ICC, cells have most or all of their extracellular matrix removed. Again, the signaling molecule may be a fluorophore or an enzyme.
The basic protocols and controls follow that of IF (above). Sample preparation, signaling excitation, and assay readout vary widely depending on individual assay conditions.
Controls for visualization methods
Visualization methods should employ positive, negative, and blank controls.
Antibody Applications - Cell Sorting Methods
Flow cytometry
Flow cytometry, used commonly in immunology, allows you to identify, analyze, capture, and sort single cells. Cells, with labeled antibodies bound to surface receptors, are “flowed” through a flow cytometer, which uses lasers to excite and read fluorophore emissions. Cells can also be sorted (separated and deposited into tubes or plate wells) by fluorescence activated cell sorting (FACS).
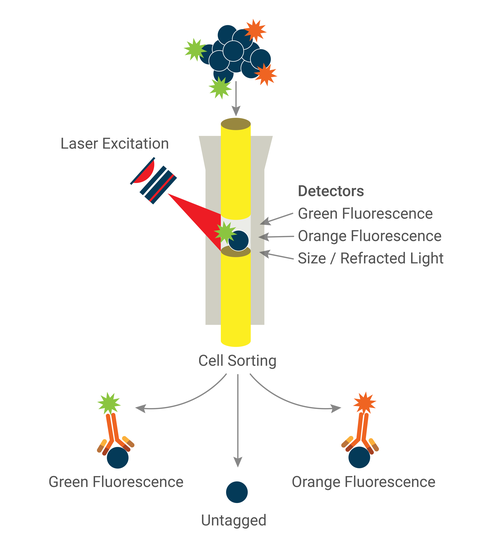
Overview of application
- Collect cells from cell culture or tissue samples.
- Wash cells, label with one or more conjugated primary antibodies, and wash again.
- Use controls to identify, or gate, cells by size and signals in the flow cytometry software.
- Flow sample cells, one at a time, through the flow cytometer, passing through the path of a laser which excites the fluorophore and reads the resulting light emissions.
- In FACS, cells are then slotted in a specific direction based on the emission(s) read by the machine.
Controls for cell sorting methods
Flow cytometry requires a negative control, a small subset of the sample with no antibodies. It also requires single-antibody controls, a small subset of the sample labeled with only one antibody, for each antibody used. Blank controls in the form of sample buffer must also be run. Unique samples (non-biological replicates) often each require their own set of controls.
Multiplex Assays
Flow cytometry, IF, IHC, and ICC are typically run as multiplex assays.
Both FACS and flow cytometers can use multiple lasers and fluorophores to distinguish between cells. Most flow experiments will use one marker to distinguish between live and dead cells, another marker to differentiate between immune and non-immune cells, and then rely on additional markers to distinguish between specific types of immune cells. Immunofluorescence experiments often use a nuclei marker to identify individual cells and additional markers to distinguish cell types.
Multiplex assays can range from two antibodies to upwards of twenty. Laser and software capacity theoretically allow for roughly fifty different fluorophore colors in flow cytometry, but the largest panels used currently are in the mid-forties. When designing a multiplex assay, selection of the fluorophores used is important. It is best to inversely pair the brightness of the fluorophore with the abundance of the antibody target. If a bright fluorophore is on an abundant target, it obscures fainter signals. Fluorophores can emit signals that overlap with each other. Choose fluorophores with minimal signaling overlap to avoid confusing data. Check that your machine has appropriate lasers and filters for each fluorophore you select.
Suggested panels for small multiplex experiments
- 4-color flow panel: BV421, FITC, PE, and APC
- 4-color microscopy panel: DAPI for nuclei, AF488, AF555, and AF647
Antibody Selection
Selecting the right antibody for your experiment is a critical step in your experimental design. Use this section to help you select antibodies for your approach.
Primary antibodies
To select an antibody, first identify your assay type and what species your protein of interest comes from. Then consider the following questions:
- Do you need polyclonal or monoclonal antibodies?
- Will you be using a direct or indirect detection method? If indirect, do you already have secondary antibodies available in your lab?
- Will you be buying conjugated antibodies or conjugating them in the lab?
- How will the sample be prepped, and will this or other factors affect epitope availability?
Once you have answered these questions, use an online resource such as Biocompare to find antibodies for your target of interest that are validated for your assay type. See the Antibody Validation section below for more information on validation.
If no antibodies are available, change the least important factor (for instance, looking for a different conjugate or looking for antibodies validated for a similar assay type) and search again.
After filtering, narrow down further by company, price, and other factors like availability and shipping time. Choose antibodies from companies that are known to produce reliable antibodies. Be aware that one antibody can and often is licensed to multiple companies for sale. Before purchasing, directly compare the product information for similar antibodies, looking for ones that have identical features and epitopes, to ensure you do not validate the same antibody multiple times.
You may end up with several suitable antibodies; if they all meet your criteria, you may either purchase several to compare, or select one based on price, shipping availability, ease of purchase, or similar factors.
Secondary antibodies
Secondary antibodies, often called secondaries, are usually much easier to select. There are three main considerations.
-
What is the source of your primary antibody?
Secondary antibodies bind to the constant region of antibodies, which is conserved by species. Therefore, secondary antibodies are species specific and are often referred to by [secondary source] [primary source secondary recognizes], e.g., “goat anti-rabbit.” Choose a secondary that recognizes your primary antibody source species. Remember that recombinant antibodies will also have a source species. - What conjugate do you want to use?
Most secondary antibodies will be available conjugated to a wide variety of signaling molecules. Select one appropriate for your application and experimental design. - Will you be using highly multiplexed visualization applications?
You may need to buy isotype-specific or even subclass-specific secondary antibodies to allow for differentiation between primary antibodies sourced from the same species.
Antibody Validation
Validating an antibody ensures that the antibody specifically recognizes and reacts to your target epitope in the assay type you are running. Sufficient validation data specific to your antibody and assay may already be published. If it is not, you’ll need to validate the antibody upon receiving it.
If an antibody fails validation, select a new antibody using the above process and try again. In some cases, it may be preferable to buy small aliquots of many different antibodies and do batch validation up front rather than risk delaying a process through sequential rounds of validation.
Validation of an antibody is a process separate from running appropriate controls in your assay. There are five types of strategies that can be used to validate antibodies, either by themselves or in combination: orthogonal, capture mass spectrometry (MS), independent antibody, genetic, and recombinant expression.
Orthogonal validation
This approach compares the results, such as protein levels, of the antibody-based assay with the results of a non-antibody assay measuring either the same output or a proxy for that output. Orthogonal validation can be done using methods such as transcriptomics or proteomics. This approach is often appropriate for quantitative methods.
Capture MS validation
This method uses peptide identification by proteomics (Capture MS) to validate the identity of proteins recognized by the antibody. It is appropriate for any assay in which the protein of interest can be isolated and analyzed. Validation relies on size comparison of peptides.
Independent antibody validation
This method validates by comparing your antibody of interest to a second, independent antibody for the same protein of interest, with no overlapping epitopes. It is not appropriate for validation of an antibody recognizing a specific epitope and may not be appropriate for polyclonal antibodies without defined epitopes.
Genetic validation
In this method, the protein of interest is knocked down or out, and antibody reactivity is compared between the wild-type and the knockdown or knockout sample. This method is appropriate for most uses but can be time and labor intensive.
Recombinant expression validation
A recombinant expression strategy is used to express the protein of interest in a cell line with low or no expression; antibody reactivity is then compared between the knock-in cell line and a cell line known to express the protein of interest. This method is appropriate for most uses, but can be time and labor intensive.

Ready to put your new knowledge to use? Addgene now makes ready-to-use recombinant antibodies made from plasmids in our collection.