CRISPR Guide
CRISPR Overview
Class 2 Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) systems, which form an adaptive immune system in bacteria, have been modified for genome engineering. Prior to CRISPR, genome engineering approaches like zinc finger nucleases (ZFNs) or transcription-activator-like effector nucleases (TALENs) required scientists to design and generate a new nuclease pair for every genomic target. Due to its comparative simplicity and adaptability, CRISPR has rapidly become the most popular genome engineering approach.

Engineered CRISPR systems contain two components: a guide RNA (gRNA or sgRNA) and a CRISPR-associated endonuclease (Cas protein). The gRNA is a short synthetic RNA composed of a scaffold sequence necessary for Cas-binding and a user-defined ∼20 nucleotide spacer that defines the genomic target to be modified. Thus, one can change the genomic target of the Cas protein by simply changing the target sequence present in the gRNA.
CRISPR was originally employed to knock out target genes in various cell types and organisms, but modifications to various Cas enzymes have extended CRISPR to selectively activate/repress target genes, purify specific regions of DNA, image DNA in live cells, and precisely edit DNA and RNA. Furthermore, the ease of generating gRNAs makes CRISPR one of the most scalable genome editing technologies. This advantage makes CRISPR perfect for genome-wide screens.
This guide will provide a basic understanding of CRISPR biology, introduce the various applications of CRISPR, and help you get started using CRISPR in your own research.
Generating a Knockout Using CRISPR

You can use CRISPR to generate knockout cells or animals by co-expressing an endonuclease like Cas9 or Cas12a (also known as Cpf1) and a gRNA specific to the targeted gene. The genomic target can be any ∼20 nucleotide DNA sequence, provided it meets two conditions:
- The sequence is unique compared to the rest of the genome.
- The target is present immediately adjacent to a Protospacer Adjacent Motif (PAM).
The PAM sequence serves as a binding signal for Cas9, but the exact sequence depends on which Cas protein you use. We'll use the popular S. pyogenes Cas9 (SpCas9) as an example, but check out our list of additional Cas proteins and PAM sequences. Once expressed, the Cas9 protein and the gRNA form a ribonucleoprotein complex through interactions between the gRNA scaffold and surface-exposed positively-charged grooves on Cas9. Cas9 undergoes a conformational change upon gRNA binding that shifts the molecule from an inactive, non-DNA binding conformation into an active DNA-binding conformation. Importantly, the spacer region of the gRNA remains free to interact with target DNA.
Cas9 will only cleave a given locus if the gRNA spacer sequence shares sufficient homology with the target DNA. Once the Cas9-gRNA complex binds a putative DNA target, the seed sequence (8-10 bases at the 3′ end of the gRNA targeting sequence) will begin to anneal to the target DNA. If the seed and target DNA sequences match, the gRNA will continue to anneal to the target DNA in a 3′ to 5′ direction. Thus, mismatches between the target sequence in the 3′ seed sequence completely abolish target cleavage, whereas mismatches toward the 5′ end distal to the PAM often still permit target cleavage.
Cas9 undergoes a second conformational change upon target binding that positions the nuclease domains, called RuvC and HNH, to cleave opposite strands of the target DNA. The end result of Cas9-mediated DNA cleavage is a double-strand break (DSB) within the target DNA (∼3-4 nucleotides upstream of the PAM sequence).
The resulting DSB is then repaired by one of two general repair pathways:
- The efficient but error-prone non-homologous end joining (NHEJ) pathway
- The less efficient but high-fidelity homology directed repair (HDR) pathway
The NHEJ repair pathway is the most active repair mechanism, and it frequently causes small nucleotide insertions or deletions (indels) at the DSB site. The randomness of NHEJ-mediated DSB repair has important practical implications, because a population of cells expressing Cas9 and a gRNA will result in a diverse array of mutations (for more information, jump to Plan Your Experiment). In most cases, NHEJ gives rise to small indels in the target DNA that result in amino acid deletions, insertions, or frameshift mutations leading to premature stop codons within the open reading frame (ORF) of the targeted gene. The ideal end result is a loss-of-function mutation within the targeted gene. However, the strength of the knockout phenotype for a given mutant cell must be validated experimentally.
Browse Plasmids: Double-Strand Break (Cut)
Enhancing Specificity with Nickases and High Fidelity Enzymes

CRISPR specificity is partially determined by how specific the gRNA targeting sequence is for the genomic target compared to the rest of the genome. Ideally, a gRNA targeting sequence will have perfect homology to the target DNA with no homology elsewhere in the genome. Realistically however, a given gRNA targeting sequence will have additional sites throughout the genome where partial homology exists. These sites are called off-targets and need to be considered when designing a gRNA for your experiment (see Plan Your Experiment below).
In addition to optimizing gRNA design, CRISPR specificity can also be increased through modifications to Cas9. As discussed previously, Cas9 generates double-strand breaks (DSBs) through the combined activity of two nuclease domains, RuvC and HNH. Cas9 nickase, a D10A mutant of SpCas9, retains one nuclease domain and generates a DNA nick rather than a DSB.
Thus, two nickases targeting opposite DNA strands are required to generate a DSB within the target DNA. This requirement for a double nick or dual nickase CRISPR system dramatically increases target specificity, since it’s unlikely that two off-target nicks will be generated close enough to cause a DSB. If high specificity is crucial to your experiment, you might consider using the dual nickase approach to create a double nick-induced DSB. The nickase system can also be combined with HDR-mediated gene editing for specific gene edits.
In 2015, researchers used rational mutagenesis to develop two high fidelity Cas9's: eSpCas9(1.1) and SpCas9-HF1. eSpCas9(1.1) contains alanine substitutions that weaken the interactions between the HNH/RuvC groove and the non-target DNA strand, preventing strand separation and cutting at off-target sites. Similarly, SpCas9-HF1 lowers off-target editing through alanine substitutions that disrupt Cas9's interactions with the DNA phosphate backbone. Another high fidelity Cas9, HypaCas9, was developed in 2017 and contains mutations in the REC3 domain that increase Cas9 proofreading and target discrimination. All three high fidelity enzymes generate less off-target editing than wild type Cas9.
Since then, another generation of high fidelity enzymes have been engineered using cell-based selection approaches. Yeast-based screening systems resulted in evolved Cas9 (evoCas9), which has four mutations in the REC3 domain. evoCas9 has less off-target activity than SpCas9-HF1 or eSpCas9(1.1). Phage-assisted continuous evolution (PACE) methods resulted in xCas9 3.7 which has 7 mutations found in the REC2, REC3, and PAM interacting domains and allows for expanded PAM recognition as well as increased specificity and lower off-target activity. By screening E. coli cells transformed with a pooled library of SpCas9 variants, researchers identified Sniper-Cas, which has less off-target activity than wild type Cas9 and is compatible with truncated gRNAs for increased specificity.
Browse Plasmids: Single-Strand Break (Nick) , Double-Strand Break (Cut)
Making Precise Modifications Using Homology Directed Repair (HDR)
While NHEJ-mediated DSB repair often disrupts the open reading frame of the gene, homology directed repair (HDR) can generate specific nucleotide changes ranging from a single nucleotide change to large insertions like the addition of a fluorophore or tag.
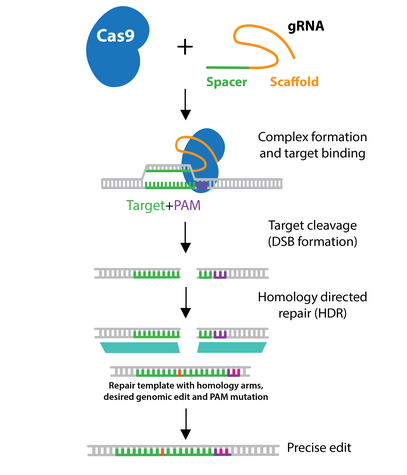
In order to utilize HDR for gene editing, a DNA repair template containing the desired sequence must be delivered into the cell type of interest with the gRNA(s) and Cas9 or Cas9 nickase. The repair template must contain the desired edit as well as additional homologous sequence immediately upstream and downstream of the target (termed left & right homology arms.) The length of each homology arm is dependent on the size of the change being introduced, with larger insertions requiring longer homology arms.
Depending on the application, the repair template may be a single-stranded oligonucleotide, double-stranded oligonucleotide, or a double-stranded DNA plasmid. When designing the repair template, do not include the PAM sequence present in the genomic DNA. This step prevents the repair template from being a suitable target for Cas9 cleavage. For example, you could alter the PAM sequence in your HDR template with a silent mutation that does not change the amino acid sequence.
The efficiency of HDR is generally low (<10% of modified alleles). For this reason, many laboratories try to enhance HDR by synchronizing the cells, since HDR takes place during the S and G2 phases of the cell cycle. Chemically or genetically inhibiting genes involved in NHEJ may also increase HDR frequency. Additionally, Cas9-CtIP, a fusion of Cas9 and CtIP, a protein involved in double-stranded break resection, can contribute to increased HDR efficiency.
Since the efficiency of Cas9 cleavage is relatively high and the efficiency of HDR is relatively low, a large portion of the Cas9-induced DSBs will be repaired via NHEJ. In other words, the resulting population of cells will contain some combination of wild type alleles, NHEJ-repaired alleles, and/or the desired HDR-edited allele. Therefore, it is important to confirm the presence of the desired edit experimentally and to isolate clones containing the desired edit (see the validation section in Plan Your Experiment).
Browse Plasmids: Endogenous Tagging
CRISPR Base Editing Without Double-Strand Breaks
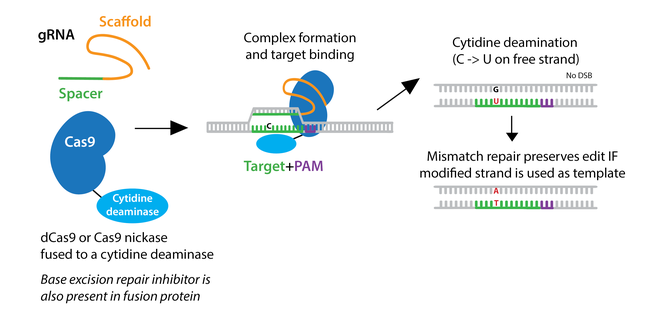
To overcome low HDR efficiency, researchers have developed two classes of base editors--cytosine base editors (CBEs) and adenine base editors (ABEs).
Cytosine base editors are created by fusing Cas9 nickase or catalytically inactive “dead” Cas9 (dCas9) to a cytidine deaminase like APOBEC. Base editors are targeted to a specific locus by a gRNA, and they can convert cytidine to uridine within a small editing window near the PAM site. Uridine is subsequently converted to thymidine through base excision repair, creating a C to T change (or a G to A on the opposite strand.)
Likewise, adenosine base editors have been engineered to convert adenosine to inosine, which is treated like guanosine by the cell, creating an A to G (or T to C) change. Adenine DNA deaminases do not exist in nature, but have been created by directed evolution of the Escherichia coli TadA, a tRNA adenine deaminase. Like cytosine base editors, the evolved TadA domain is fused to a Cas9 protein to create the adenine base editor.
Both types of base editors are available with multiple Cas9 variants including high fidelity Cas9’s. Further advancements have been made by optimizing expression of the fusions, modifying the linker region between Cas variant and deaminase to adjust the editing window, or adding fusions that increase product purity such as the DNA glycosylase inhibitor (UGI) or the bacteriophage Mu- derived Gam protein (Mu- GAM).
While many base editors are designed to work in a very narrow window proximal to the PAM sequence, some base editing systems create a wide spectrum of single-nucleotide variants (somatic hypermutation) in a wider editing window, and are thus well suited to directed evolution applications. Examples of these base editing systems include targeted AID-mediated mutagenesis (TAM) and CRISPR-X, in which Cas9 is fused to activation-induced cytidine deaminase (AID).
Browse Plasmids: Base Edit
Other CRISPR systems, specifically the Type VI CRISPR enzymes Cas13a/C2c2 and Cas13b, target RNA rather than DNA. Fusing a hyperactive adenosine deaminase that acts on RNA, ADAR2(E488Q), to catalytically dead Cas13b creates a programmable RNA base editor that converts adenosine to inosine in RNA (termed REPAIR). Since inosine is functionally equivalent to guanosine, the result is an A->G change in RNA. The catalytically inactive Cas13b ortholog from Prevotella sp., dPspCas13b, does not appear to require a specific sequence adjacent to the RNA target, making this a very flexible editing system. Editors based on a second ADAR variant, ADAR2(E488Q/T375G), display improved specificity, and editors carrying the delta-984-1090 ADAR truncation retain RNA editing capabilities and are small enough to be packaged in AAV particles.

Browse Plasmids: RNA Editing
Activation or Repression of Target Genes Using CRISPR
CRISPR is a remarkably flexible tool for genome manipulation, since Cas enzymes bind target DNA independently of their ability to cleave target DNA. Specifically, both RuvC and HNH nuclease domains can be rendered inactive by point mutations (D10A and H840A in SpCas9), resulting in a nuclease dead Cas9 (dCas9) molecule that cannot cleave target DNA. The dCas9 molecule retains the ability to bind to target DNA based on the gRNA targeting sequence.
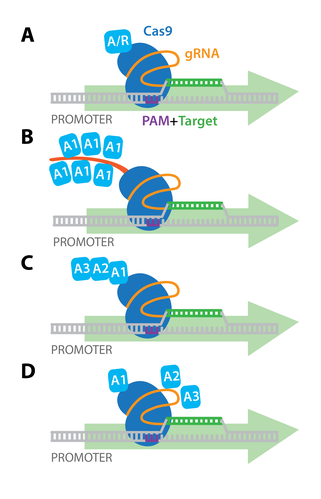
Early experiments demonstrated that targeting dCas9 to transcription start sites was sufficient to repress transcription by blocking initiation. dCas9 can also be fused with transcriptional repressors or activators, and targeting these dCas9 fusion proteins to the promoter region results in robust transcriptional repression (CRISPR interference, or CRISPRi) or activation (CRISPRa) of downstream target genes. The simplest dCas9-based activators and repressors consist of dCas9 fused directly to a single transcriptional activator (e.g. VP64) or repressor (e.g. KRAB; see panel A to the right).
Additionally, more elaborate activation strategies have been developed for more potent activation of target genes in mammalian cells. These include: co-expression of epitope-tagged dCas9 and antibody-activator effector proteins (e.g. SunTag system , panel B); dCas9 fused to several different activation domains in series (e.g. dCas9-VPR, panel C); or co-expression of dCas9-VP64 with a modified scaffold gRNA and additional RNA-binding helper activators (e.g. SAM activators, panel D). Importantly, unlike the genome modifications induced by Cas9 or Cas9 nickase, dCas9-mediated gene activation or repression is reversible, since it does not permanently modify the genomic DNA. dCas9 has also been used for genome-wide screens to activate or repress gene expression in mice and human cells.
In the bacterial realm, activating gene expression is more difficult because of the absence of effective gene activators when fused with molecular DNA binding domains such as dCas9. Recently, synthetic CRISPR-Cas gene activators have been developed for bacteria by using a scaffold RNA that contains the gRNA and an RNA hairpin to recruit activation proteins.
CRISPRi has also been adapted for diverse bacterial species using a modular system called Mobile-CRISPRi. In this system, CRISPRi is introduced into bacteria using conjugation and stably integrated into the chromosome.
Browse Plasmids: Activate, Repress/Interfere
Epigenetic Modifications Using CRISPR
Inactive Cas enzymes can be fused to epigenetic modifiers like p300, LSD1, MQ1, and TET1 to create programmable epigenome-engineering tools. These tools alter gene expression without inducing double-strand breaks by modifying the methylation state of cytosines in a gene’s promoter or by inducing histone acetylation or demethylation. CRISPR epigenetic tools are specific for particular chromatin and DNA modifications, allowing researchers to isolate the effects of a single epigenetic mark.
Another potential advantage of CRISPR epigenetic tools is their persistence and inheritance. CRISPR activators and repressors are thought to be reversible once the effector is inactivated or removed from the system. In contrast, epigenetic marks left by targeted epigenetic modifiers may be more frequently inherited by daughter cells. In certain cases, epigenetic modifiers may work better than activators/repressors in modulating transcription. However, since the effects of these tools are likely cell type- and context-dependent, it might be beneficial to try multiple CRISPR strategies when setting up your experimental system.
Browse Plasmids: Epigenetics

Multiplex Genome Engineering with CRISPR
Expressing several gRNAs from the same plasmid ensures that each cell containing the plasmid will express all of the desired gRNAs, thus increasing the likelihood that all desired genomic edits will be carried out by Cas9. Such multiplex CRISPR applications include:
- The use of dual nickases to generate a knockout or gene edit
- Using Cas9 to generate large genomic deletions by removing the sequence between two gRNA target sites
- Modifying multiple genes at once
Current multiplex CRISPR systems enable researchers to target anywhere from 2 to 7 genetic loci by cloning multiple gRNAs into a single plasmid. These multiplex gRNA vectors can conceivably be combined with any of the aforementioned CRISPR derivatives to knock out, activate, or repress target genes. Read more about Cas9 multiplexing and Cas12a (Cpf1) multiplexing.
Browse Plasmids: Multiplex gRNA vectors
Genome-Wide Screens Using CRISPR
The ease of gRNA design and synthesis, as well as the ability to target almost any genomic locus, make CRISPR the ideal genome engineering system for large-scale forward genetic screening. Forward genetic screens are particularly useful for studying diseases or phenotypes for which the underlying genetic cause is not known. In general, the goal of a genetic screen is to generate a large population of cells with mutations in, or activation/repression of, a wide variety of genes and then use these cells to identify the genetic perturbations that result in a desired phenotype.
Before CRISPR, genetic screens relied heavily on shRNA technology, which is prone to off-target effects and false negatives due to incomplete knockdown of target genes. In contrast, CRISPR is capable of making highly specific, permanent genetic modifications that are more likely to ablate target gene function. CRISPR has already been used extensively to screen for novel genes that regulate known phenotypes, including resistance to chemotherapy drugs, resistance to toxins, cell viability, and tumor metastasis. Currently, the most popular method for conducting genome-wide screens using CRISPR involves the use of pooled lentiviral CRISPR libraries.
What are pooled lentiviral CRISPR libraries?
Pooled lentiviral CRISPR libraries (often referred to as CRISPR libraries) are a heterogenous population of lentiviral transfer vectors, each containing an individual gRNA targeting a single gene in a given genome.
Guide RNAs are designed in silico and synthesized (see panel A below), then cloned in a pooled format into lentiviral transfer vectors (panel B). CRISPR libraries have been designed for common CRISPR applications including genetic knockout, activation, and repression for human and mouse genes.

Each CRISPR library is different, as libraries can target anywhere from a single class of genes to every gene in the genome. However, there are several features that are common across most CRISPR libraries. First, each library typically contains ∼3-6 gRNAs per gene to ensure modification of every target gene, so CRISPR libraries contain thousands of unique gRNAs targeting a wide variety of genes. gRNA design for CRISPR libraries is usually optimized to select for gRNAs with high on-target activity and low off-target activity, and libraries may use different algorithms for gRNA design.
Keep in mind that the exact region of the gene to be targeted varies depending on the specific application. For example, knockout libraries often target 5′ constitutively expressed exons, but activation and repression libraries will target promoter or enhancer regions. Be sure to check the library information/original publication to see if a library is suitable for your experiment. Libraries may be available in a 1-plasmid system, in which Cas9 is included on the gRNA-containing plasmid, or a 2-plasmid system in which Cas9 must be delivered separately.
Although lentiviral libraries containing Cas9 are the most popular method for CRISPR screening, they are not suitable for all cell types or experiments. Mammalian CRISPR libraries have also been created in AAV backbones for in vivo experiments and in a retroviral backbone for delivery to cells that are poorly transduced by lentivirus. Non-mammalian CRISPR libraries are also available. Additionally, although CRISPR has been less widely used in bacteria due to technical challenges, several bacterial CRISPR libraries have been developed for inhibition using dCas9.
How do you use a CRISPR library?
CRISPR libraries from Addgene are available in two formats: as DNA, or in select cases, as pre-made lentivirus.
In the case of DNA libraries, the CRISPR library will be shipped at a concentration that is too low to be used in experiments. Thus, the first step in using your library is to amplify the library (panel C) to increase the total amount of DNA using the protocol specified by the depositing lab. When amplifying the library, it is important to maintain good representation of gRNAs so that the composition of your amplified library matches that of the original library. You'll use next-generation sequencing (NGS) to verify that this is the case. Learn more about library verification.
Once the library has been amplified/verified, the next step is to generate lentivirus containing the entire CRISPR library (panel D). Then, you will transduce cells with the lentiviral library (panel E). Remember - if you are using a 2-vector system, you will need to transduce cells that are already expressing Cas9. After applying your screening conditions, you will look for relevant genes (hits) using NGS technology. For more detail on using CRISPR for both positive and negative screens, see our pooled library guide.
What can screens tell you?
As noted above, forward genetic screens are most useful for situations in which the physiology or cell biology behind a particular phenotype or disease is well understood, but the underlying genetic causes are unknown. Therefore, genome-wide screens using CRISPR libraries are a great way to gather unbiased information regarding which genes play a causal role in a given phenotype. With any experiment, it is important to verify that the hits you identify are actually important for your phenotype of interest. You can individually test the gRNAs identified in your screen to ensure that they reproduce the phenotype of interest.
Find more information on factors to consider before starting your pooled library experiment in “Practical Considerations for Using Pooled Lentiviral CRISPR Libraries” (McDade et al., 2016).
Browse Libraries: CRISPR Pooled Libraries
Visualize Genomic Loci Using Fluorophores
Using catalytically inactive dCas9 fused to a fluorescent marker like GFP, researchers have turned dCas9 into a customizable DNA labeler compatible with fluorescence microscopy in living cells. Alternatively, gRNAs can be fused to protein-interacting RNA aptamers, which recruit specific RNA-binding proteins (RBPs) tagged with fluorescent proteins to visualize targeted genomic loci.
CRISPR imaging has numerous advantages over other imaging techniques, including that it is easy to implement due to the simplicity of gRNA design, programmable for different genomic loci, capable of detecting multiple genomic loci, and compatible with live cell imaging. Compared to techniques like fluorescence in situ hybridization (FISH), CRISPR imaging offers a unique method for detecting the chromatin dynamics in living cells.
Multicolor CRISPR imaging allows for simultaneous tracking of multiple genomic loci in living cells. One method uses orthogonal dCas9’s (e.g., S. pyogenes dCas9 and S. aureus dCas9) tagged with different fluorescent proteins. Another method uses gRNAs fused to orthogonal protein-interacting RNA aptamers, which recruit specific orthogonal RBPs tagged with different fluorescent proteins, (CRISPRainbow kit). It has been shown that gRNAs carrying 8 aptamers (CRISPR-Sirius), provide a better stability and enable a signal amplification for better imaging of genomic loci.
The fluorescent CRISPR system has been used for dynamic tracking of repetitive and non-repetitive genomic loci, as well as chromosome painting in living cells. Visualizing a specific genomic locus requires recruitment of many copies of labeled proteins to the given region. For example, chromosome-specific repetitive loci can be efficiently visualized in living cells using a single gRNA that has multiple targeted sequences in close proximity. A non-repetitive genomic locus can also be labeled by co-delivering multiple gRNAs that tile the locus. Chromosome painting requires delivery of hundreds of gRNAs with target sites throughout the chromosome.

Browse Plasmids: Visualize
Purify Genomic Regions Using dCas9
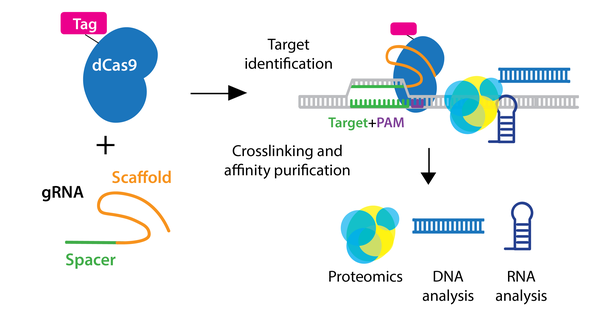
Identifying molecules associated with a genomic region of interest in vivo is essential to understanding locus function. Using CRISPR, researchers have expanded chromatin immunoprecipitation (ChIP) to allow purification of any genomic sequence specified by a particular gRNA.
In the enChIP (engineered DNA-binding molecule-mediated ChIP) system, catalytically inactive dCas9 is used to purify genomic DNA bound by the gRNA. An epitope tag(s) can be fused to dCas9 or gRNA for efficient purification. Various epitope tags including 3xFLAG-tag, PA, and biotin tags, can be used for enChIP, as well as an anti-Cas9 antibody. Biotin tagging of dCas9 can be achieved by fusing a biotin acceptor site to dCas9 and co-expressing BirA biotin ligase, as seen in the CAPTURE system. The locus is subsequently isolated by affinity purification against the epitope tag.
After purification of the locus, molecules associated with the locus can be identified by mass spectrometry (proteins), RNA-sequencing (RNAs), and NGS (other genomic regions). Compared to conventional methods for genomic purification, CRISPR-based purification methods are more straightforward and enable direct identification of molecules associated with a genomic region of interest in vivo.
Browse Plasmids: Purify
RNA Targeting
Type VI CRISPR enzymes such as Cas13 recognize ssRNA rather than dsDNA. Many of these enzymes also have the ability to process crRNA precursors to mature crRNAs. Upon ssRNA recognition by the crRNA, the target RNA is degraded. In bacteria, Cas13 enzymes can also cleave RNAs non-specifically after the initial crRNA-guided cleavage. This promiscuous cleavage activity slows bacterial cell growth and may further protect bacteria from viral pathogens. Based on this feature, a Cas13a-based molecular detection platform, termed SHERLOCK, has been used to differentiate strains of Zika virus, genotype human DNA, and identify tumor mutations within cell-free genomic DNA. This non-specific cleavage does not occur in mammalian cells. Similar to Cas9 and Cas12 , Cas13 can be converted to an RNA-binding protein through mutation of its catalytic domain. Fusions to catalytically inactive Cas13 can be used to modulate RNA editing, for in vivo RNA imaging, or to control alternative splicing. Learn more about Cas13a or Cas13d.
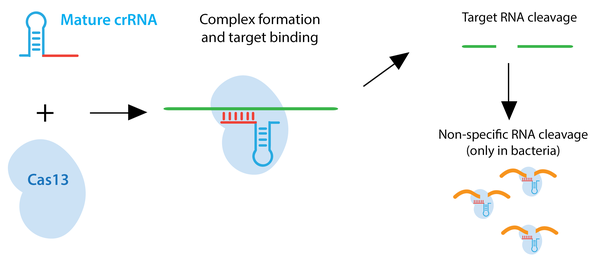
Browse Plasmids: RNA Targeting
Cas9 Alternatives for CRISPR Genome Engineering
While S. pyogenes Cas9 (SpCas9) is certainly the most commonly used CRISPR endonuclease for genome engineering, it may not be the best endonuclease for every application. For example, the PAM sequence for SpCas9 (5′-NGG-3′) is abundant throughout the human genome, but an NGG sequence may not be positioned correctly to target your desired genes for modification. This limitation is of particular concern when trying to edit a gene using homology directed repair (HDR), which requires PAM sequences in very close proximity to the region to be edited.
To address these limitations, researchers have engineered SpCas9 enzymes with altered PAM specificities using a variety of approaches including phage-assisted evolution and directed mutagenesis. This resulted in the development of several SpCas9-derived variants with non-NGG PAM sequences. Another Cas9 alternative is xCas9, which targets a broad set of PAM sequences, such as NG, GAA, and GAT, while also displaying minimal off-target activity. SpCas9-NG, a variant that recognizes the NG PAM, has increased activity in vitro relative to other Cas9 endonucleases. Read more about Cas9 variants.
Additional Cas9 orthologs from various species bind a variety of PAM sequences. These enzymes may have other characteristics that make them more useful than SpCas9 for specific applications. For example, the relatively large size of SpCas9 (∼4 kb coding sequence) means that plasmids carrying the SpCas9 cDNA cannot be efficiently packaged into adeno-associated virus (AAV). Since the coding sequence for Staphylococcus aureus Cas9 (SaCas9) is ∼1 kb shorter than SpCas9, SaCas9 can be efficiently packaged into AAV. Similar to SpCas9, the SaCas9 endonuclease is capable of modifying target genes in mammalian cells in vitro and in mice in vivo.
Another limitation of SpCas9 is the low efficiency of making specific genetic edits via HDR. For specific point edits, CRISPR base editing is a useful alternative to HDR. For larger edits, Cas12a (Cpf1) may be a better option. Unlike Cas9 nucleases, which create blunt DSBs, Cas12a-mediated DNA cleavage creates DSBs with a short 3′ overhang. Cas12a’s staggered cleavage pattern opens up the possibility of directional gene transfer, analogous to traditional restriction enzyme cloning, which may increase the efficiency of gene editing. Like the Cas9 variants and orthologs described above, Cas12a also expands the range of sites that can be targeted by CRISPR to AT-rich regions or AT-rich genomes that lack the NGG PAM sites favored by SpCas9. Similar to Cas9, Cas12a has also been engineered to recognize different PAM variants.
Want more information on the wide variety of Cas enzymes? CasPEDIA is an encyclopedia of Class 2 CRISPR systems with wiki entries describing enzyme activity, experimental considerations, and more. CasPEDIA is a community resource created and maintained by the Doudna Lab at the Innovative Genomics Institute, University of California, Berkeley.
Anti-CRISPR
In order to limit off-target effects of CRISPR-Cas9, researchers use a class of small proteins called Acr (Anti-CRISPR) proteins to control CRISPR activity. Acr proteins are found in phages that have evolved ways to overcome the endogenous CRISPR systems used by various bacteria and archaea to protect against invading nucleic acids, such as phage genomes. Because of this diversity, Acr family members inhibit CRISPR by a variety of mechanisms. Some Acr proteins interfere with DNA binding at the PAM site whereas others interfere with the Cas9 HNH endonuclease domain. Furthermore, Acr proteins can be specific for a particular Cas9 species or can inhibit CRISPR enzymes across multiple bacterial species. For example, AcrIIA2 and AcrIIA4 isolated from Listeria monocytogenes can inhibit CRISPR activity of both LmoCas9 and SpCas9.
Acr proteins are a great way to control when genome editing can happen. For example, Dominik Niopek’s lab developed a way to optogenetically control genome editing by fusing the anti-CRISPR protein AcrIIA4 with the photo-sensitive LOV2 domain. This allows researchers to switch genome editing on and off using light.
Browse Plasmids: Anti-CRISPR
Plan Your CRISPR Experiment
Get Started
CRISPR is a powerful system that enables researchers to manipulate the genome like never before. This section will provide a general framework to get you started using CRISPR in your research. Although we will use the example of CRISPR/Cas9 in mammalian cells, many of these principles apply to using CRISPR in other organisms. First, consider the genetic manipulation that is necessary to model your specific disease or process of interest. Do you want to:
- Generate complete and permanent loss of gene expression or function (knockout)?
- Generate a specific mutant allele of a gene (point mutant)?
- Increase or decrease expression of a target gene?
Once you have a clear understanding of your experimental goal, you are ready to start navigating the different reagents that are available for your particular experiment.
Select Your Desired Genetic Manipulation
Different genetic manipulations require different CRISPR components. Selecting a specific genetic manipulation can be a good way to narrow down which reagents are appropriate for a given experiment. Make sure to check whether reagents are available to carry out your experiment in your particular model organism. There may not be a perfect plasmid for your specific application, and in such cases, it may be necessary to customize an existing reagent to suit your needs.
| Genetic Manipulation | Application | Cas9 | gRNA | Additional Considerations |
|---|---|---|---|---|
| Knockout | Permanently disrupt gene function in a particular cell type or organism without a specific preferred mutation | Cas9 (or Cas9 nickase) | Single (or dual) gRNA targeting 5′ exon or essential protein domains | High-fidelity Cas enzymes increase specificity. Dual-nickase approach increases specificity but is less efficient. Each putative knockout allele must be experimentally verified. |
| Edit | Generate a specific user-defined sequence change in a particular gene, such as generating a point mutation or inserting a tag | Cas9 (or Cas9 nickase); Base editor | Single (or dual) gRNA targeting the region where the edit should be made | HDR requires a repair template and displays reduced efficiency compared to NHEJ knockout. Base editors can make a limited set of mutations. |
| Repress or Interfere (CRISPRi) | Reduce expression of a particular gene(s) without permanently modifying the genome | dCas9-repressor (such as dCas9-KRAB) or dCas9 | gRNA(s) targeting promoter elements of target gene | dCas9-KRAB is more effective than dCas9 alone for mammalian cell lines. |
| Activate (CRISPRa) | Increase expression of an endogenous gene(s) without permanently modifying the genome | dCas9-activator (such as dCas9-VP64) | gRNA(s) targeting promoter elements of target gene | Many different activators exist, including the multi-plasmid SAM system. |
Select Expression System
To use CRISPR, you will need both Cas9 and a gRNA expressed in your target cells. For easy-to-transfect cell types (e.g. HEK293 cells), transfection with standard transfection reagents may be sufficient to express the CRISPR machinery. For more difficult cells (e.g. primary cells), viral delivery of CRISPR reagents may be more appropriate. In cases where off-target editing is a major concern, Cas9-gRNA ribonucleoprotein (RNP) complexes are advantageous due to the transient Cas9 expression.
The table below summarizes the major expression systems and variables for using CRISPR in mammalian cells. Some of the variables include:
- Species of Cas enzyme and gRNA
- Species of promoter and expression pattern of promoter for Cas enzyme and gRNA
- Presence of a selectable marker (drug or fluorophore)
- Delivery method
| Expression System | Components of System | Application |
|---|---|---|
| Mammalian expression vector |
|
Transient or stable expression of Cas9 and/or gRNA in a mammalian cell line that can be transfected at high efficiency |
| Lentiviral transduction |
|
|
| AAV transduction |
|
|
| RNA delivery of Cas9 and gRNA | In vitro transcription reactions generate mature Cas9 mRNA and gRNA, which are then delivered to target cells through microinjection or electroporation |
|
| Cas9-gRNA ribonucleoprotein (RNP) complexes | Purified Cas9 protein and in vitro transcribed gRNA are combined to form a Cas9-gRNA complex, which is delivered to cells using cationic lipids |
|
Additional Resources:
- CRISPR protocols from Addgene depositors
- JOVE Video Reprint: Protocol for genomic deletions in mammalian cell lines
Select Your Target Sequence and Design Your gRNA
Once you have selected your CRISPR components and method of delivery, you are ready to select a target sequence and design your gRNA.
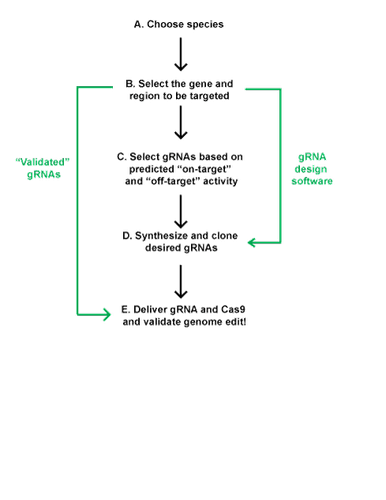
- Know your cell line/organism and genomic sequence
When possible, you should sequence the region you are planning to modify prior to designing your gRNA, as sequence variation between your gRNA targeting sequence and target DNA may result in reduced cleavage. The number of alleles for each gene may vary depending on the specific cell line or organism, which may affect the observed efficiency of CRISPR knockout or knockin.
- Select gene and genetic element to be manipulated
In order to manipulate a given gene using CRISPR, you will have to identify the genomic sequence for the gene you are trying to target. However, the exact region of the gene you target will depend on your specific application. For example:
- To activate or repress a target gene using dCas9-activators or dCas9-repressors, gRNAs should be targeted to the promoter driving expression of your gene of interest.
- For genetic knockouts, gRNAs commonly target 5′ constitutively expressed exons, which reduces the chances that the targeted region is removed from the mRNA due to alternative splicing. Exons near the N-terminus are targeted since frameshift mutations here increase the likelihood that a nonfunctional protein product is produced.
- Alternatively, gRNAs can be designed to target exons that code for known essential protein domains. The benefit of this approach is that even non-frameshift alleles may alter protein function when they occur in important protein domains.
- For gene editing experiments using HDR, it is essential that the target sequence be very close to the location of the desired edit, ideally less than 10 bp away. In this case, it is necessary to identify the exact location where the edit should occur and select a target sequence nearby.
-
Select gRNAs based on predicted on-target and off-target activity
A PAM sequence is absolutely necessary for Cas9 to bind target DNA. As such, one can start by identifying all PAM sequences within the genetic region to be targeted. If there are no PAM sequences for your chosen enzyme within your desired sequence, you may want to consider alternative Cas enzymes (see Cas9 variants and PAM sequences). Once possible PAM sequences and putative target sites have been identified, it is time to choose which site is likely to result in the most efficient on-target cleavage.
The gRNA target sequence needs to match the target locus, but it is also critical to ensure that the gRNA target sequence does NOT match additional sites within the genome. In a perfect world, your gRNA target sequence would have perfect homology to your target with no homology elsewhere in the genome. Realistically, a given gRNA target sequence will have partial homology to additional sites throughout the genome. These sites are called off-targets and should be examined during gRNA design. In general, off-target sites are not cleaved as efficiently when mismatches occur near the PAM sequence, so gRNAs with no homology or those with mismatches close to the PAM sequence will have the highest specificity. To increase specificity, you can also consider using a high-fidelity Cas enzyme.
In addition to off-target activity, it is also important to consider factors that maximize cleavage of the desired target sequence or on-target activity. Two gRNA targeting sequences with 100% homology to their DNA targets may not result in equivalent cleavage efficiency. In fact, cleavage efficiency may increase or decrease depending upon the specific nucleotides within the selected target sequence. For example, gRNA targeting sequences containing a G nucleotide at position 20 (1 bp upstream of the PAM) may be more efficacious than gRNAs containing a C nucleotide at the same position in spite of being a perfect match for the target sequence.
Many gRNA design programs can locate potential PAM and target sequences and rank the associated gRNAs based on their predicted on-target and off-target activity (see gRNA design software). Additionally, many plasmids containing validated gRNAs are now available through Addgene. These plasmids contain gRNAs that have been used successfully in genome engineering experiments. Using validated gRNAs can save your lab valuable time and resources when carrying out CRISPR experiments. Read more about how to design your gRNA.
Browse Plasmids: gRNAs
- Synthesize and clone desired gRNAs
Once you have selected your target sequences it is time to design your gRNA oligos and clone these oligos into your desired vector. In many cases, targeting oligos are synthesized, annealed, and inserted into plasmids containing the gRNA scaffold using standard restriction-ligation cloning. However, the exact cloning strategy will depend on the gRNA vector you have chosen, so it is best to review the protocol associated with the specific plasmid in question (see CRISPR protocols from Addgene depositors).
- Deliver Cas9 and gRNA
Choose a delivery method that is compatible with your experimental system. CRISPR efficiency will vary based on the method of delivery and the cell type. Before proceeding with your experiment, it may be necessary to optimize your delivery conditions. Learn more about CRISPR delivery in mammalian systems.
- Validate genetic modification
Once you have successfully delivered the gRNA and Cas enzyme to your target cells, it is time to validate your genome edit. CRISPR editing produces several possible genotypes within the resulting cell population. Some cells may be wild type due to either (1) a lack of gRNA and/or Cas9 expression or (2) a lack of efficient target cleavage in cells expressing both Cas9 and gRNA.
Edited cells may be homozygous or heterozygous for edits at your target locus. Furthermore, in cells containing two mutated alleles, each mutated allele may be different owing to the error-prone nature of NHEJ. In HDR gene editing experiments, most mutated alleles will not contain the desired edit, as a large percentage of DSBs are still repaired by NHEJ.
How do you determine that your desired edit has occurred? The exact method necessary to validate your edit will depend upon your specific application. However, there are several common ways to verify that your cells contain your desired edit, including but not limited to:
- Mismatch-cleavage assay (for NHEJ repaired DSBs): Provides a semi-quantitative readout of the percentage of alleles that have been mutated within a mixed cell population. The region of interest is PCR amplified, PCR products are denatured-renatured, treated with a nuclease that cleaves DNA heteroduplexes, and run on an agarose gel to identify DNA fragments.
- PCR and restriction digest (for HDR repaired DSBs): For small nucleotide edits that introduce a novel restriction site. The region of interest is PCR amplified, digested with the appropriate restriction enzyme and run on an agarose gel to identify DNA fragments.
- PCR amplification and gel electrophoresis (for HDR or NHEJ): For large deletions or insertions, the region of interest can be PCR amplified using primers that (A) flank the region of interest (deletions or small insertions) or (B) span the genome-insert boundary (insertions only). The PCR product is then run on an agarose gel to determine whether the edit was successful.
- PCR amplification, subcloning and Sanger sequencing (for HDR or NHEJ): Provides semi-quantitative assessment of targeting frequency and exact sequence of targeted alleles. Involves PCR amplification of targeted region from DNA, subcloning into a plasmid, and screening individual clones.
- PCR amplification and next-generation sequencing (for HDR or NHEJ): Provides quantitative assessment of the genome edits in your target sequence and can also be used to examine off-target effects.
More information on each of these techniques can be found in our blog post CRISPR 101: Validating your Genome Edit.
Resources
Addgene Website & Blog References
Website
- CRISPR plasmids and resources at Addgene
- CRISPR 101 eBook (2nd edition)
- CRISPR software and depositor protocols
- JOVE video reprint: Protocol for genomic deletions in mammalian cell lines
- Viral service at Addgene
- Viral plasmids and resources at Addgene
Blog
- CRISPR topic page
- How to design your gRNA for CRISPR genome editing
- CRISPR 101: Homology directed repair
- CRISPR 101: Validating your genomic edit
- CRISPR 101: Multiplex expression of gRNAs
- Single Base Editing with CRISPR
- CasPEDIA: A Functional Classification of Cas Enzymes
Cas Enzymes and PAM Sequences
| Species/Variant of Cas9 | PAM Sequence* |
|---|---|
| Streptococcus pyogenes (SP); SpCas9 | 3' NGG |
| SpCas9 D1135E variant | 3' NGG (reduced NAG binding) |
| SpCas9 VRER variant | 3' NGCG |
| SpCas9 EQR variant | 3' NGAG |
| SpCas9 VQR variant | 3' NGAN or NGNG |
| xCas9 | 3' NG, GAA, or GAT |
| SpCas9-NG | 3' NG |
| Staphylococcus aureus (SA); SaCas9 | 3' NNGRRT or NNGRR(N) |
| Acidaminococcus sp. (AsCpf1) and Lachnospiraceae bacterium (LbCpf1) | 5' TTTV |
| AsCpf1 RR variant | 5' TYCV |
| LbCpf1 RR variant | 5' TYCV |
| AsCpf1 RVR variant | 5' TATV |
| Campylobacter jejuni (CJ) | 3' NNNNRYAC |
| Neisseria meningitidis (NM) | 3' NNNNGATT |
| Streptococcus thermophilus (ST) | 3' NNAGAAW |
| Treponema denticola (TD) | 3' NAAAAC |
| Additional Cas9s from various species | PAM sequence may not be characterized |
*In the table above, 3' and 5' indicate on which end of targeted sequence the PAM is located. The majority of the CRISPR plasmids in Addgene’s collection are from S. pyogenes unless otherwise noted.
Glossary
| Term | Definition |
|---|---|
| Base editor | Fusion of a Cas protein to a deaminase that enables direct base conversion in RNA or DNA without a DNA double-strand break |
| Cas | CRISPR Associated Protein, includes nucleases like Cas9 and Cas12a (also known as Cpf1) |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat, a bacterial genomic region used in pathogen defense |
| CRISPRa | CRISPR Activation; using a dCas9 or dCas9-activator with a gRNA to increase transcription of a target gene |
| CRISPRi | CRISPR Interference; using a dCas9 or dCas9-repressor with a gRNA to repress/decrease transcription of a target gene |
| Cut | A DNA double-strand break; the wild type function of most Cas nucleases |
| dCas9 | Nuclease dead Cas9, an enzymatically inactive form of Cas9; can bind, but cannot cleave DNA |
| DSB | Double-Strand Break, a break in both strands of DNA |
| Dual nickase/Double nick | A method to decrease off-target effects by using a single Cas9 nickase and 2 different gRNAs, which bind in close proximity on opposite strands of the DNA, to create a DSB |
| enChIP | Engineered DNA-binding molecule-mediated ChIP, using a tagged-dCas9+gRNA to purify specific genomic regions to identify molecules associated with the genomic regions |
| Genetic modification or manipulation | Any genetic perturbation, including genetic knockout, gene activation, or gene repression |
| gRNA | Guide RNA, a synthetic fusion of the endogenous bacterial crRNA and tracrRNA that provides both targeting specificity and scaffolding/binding ability for Cas9 nuclease. This synthetic fusion does not exist in nature and is also commonly referred to as an sgRNA. |
| gRNA scaffold sequence | The sequence within the gRNA that is responsible for Cas9 binding, it does not include the 20 bp spacer/targeting sequence that is used to guide Cas9 to target DNA |
| gRNA targeting sequence | The 20 nucleotides that precede the PAM sequence in the genomic DNA. This sequence is cloned into a gRNA expression plasmid but does NOT include the PAM sequence or the gRNA scaffold sequence |
| HDR | Homology Directed Repair, a DNA repair mechanism that uses a template to repair DNA nicks or DSBs |
| Indel | Insertion/deletion, a type of mutation that can result in the disruption of a gene by shifting the ORF and/or creating premature stop codons |
| NHEJ | Non-Homologous End Joining; A DNA repair mechanism that often introduces indels |
| Nick | A break in only one strand of dsDNA |
| Nickase | Cas9 with one of the two nuclease domains inactivated. This enzyme is capable of cleaving only one strand of target dsDNA. |
| Off-target effects or off-target activity | Cas9 cleavage at undesired locations due to gRNA targeting sequence with sufficient homology to recruit Cas9 to unintended genomic locations |
| On-target activity | Cas9 cleavage at a desired location specified by a gRNA target sequence |
| ORF | Open Reading Frame; The translated codons that make up a gene |
| PAM | Protospacer Adjacent Motif; Sequence adjacent to the target sequence that is necessary for Cas enzymes to bind target DNA |
| PCR | Polymerase Chain Reaction; Used to amplify a specific sequence of DNA |
| Target locus | Genomic target of the gRNA. The sequence includes the unique ~20 bp target specified by the gRNA plus the genomic PAM sequence |
Publications
2019
- Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. 2019. Peters JM, Koo BM, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS. Nat Microbiol. Feb;4(2):244-250. PMID: 30617347
2018
- A highly specific SpCas9 variant is identified by in vivo screening in yeast. 2018. Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, Lorenzin F, Prandi D, Romanel A, Demichelis F, Inga A, Cereseto A. Nat Biotechnol. Mar;36(3):265-271. PMID: 29431739
- Base editing: precision chemistry on the genome and transcriptome of living cells. 2018. Rees HA, Liu DR. Nat Rev Genet.. Dec;19(12):770-788. PMID: 30323312
- CRISPR base editors: genome editing without double-stranded breaks. 2018. Eid A, Alshareef S, Mahfouz MM. Biochem J. Jun 11;475(11):1955-1964. PMID: 29891532
- CRISPR-Based Technologies: Impact of RNA-Targeting Systems. 2018. Terns MP. Mol Cell. Nov 1;72(3):404-412. PMID: 30388409
- CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging.. 2018. Ma H, Tu LC, Naseri A, Chung YC, Grunwald D, Zhang S, Pederson T.. Nat Methods. Nov;15(11):928-931. PMID: 30377374
- CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. 2018. Charpentier M, Khedher AHY, Menoret S, Brion A, Lamribet K, Dardillac E, Boix C, Perrouault L, Tesson L, Geny S, De Cian A, Itier JM, Anegon I, Lopez B, Giovannangeli C, Concordet JP. Nat Commun. Mar 19;9(1):1133. PMID: 29556040
- Directed evolution of CRISPR-Cas9 to increase its specificity. 2018. Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim YH, Lee K, Jung I, Kim D, Kim S, Kim JS. Nat Commun. Aug 6;9(1):3048. PMID: 30082838
- Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. 2018. Bubeck F, Hoffmann MD, Harteveld Z, Aschenbrenner S, Bietz A, Waldhauer MC, Börner K, Fakhiri J, Schmelas C, Dietz L, Grimm D, Correia BE, Eils R, Niopek D. Nat Methods. Nov;15(11):924-927. PMID: 30377362
- Engineered CRISPR-Cas9 nuclease with expanded targeting space. 2018. Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, Noda T, Abudayyeh OO, Gootenberg JS, Mori H, Oura S, Holmes B, Tanaka M, Seki M, Hirano H, Aburatani H, Ishitani R, Ikawa M, Yachie N, Zhang F, Nureki O. Science. Sep 21;361(6408):1259-1262. PMID: 30166441
- Evolved Cas9 variants with broad PAM compatibility and high DNA specificity.. 2018. Hu JH, Miller SM, Geurts MH, Tang W, Chen L, Sun N, Zeina CM, Gao X, Rees HA, Lin Z, Liu DR. Nature. Apr 5;556(7699):57-63. PMID: 29512652
- Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. 2018. Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D. PLoS Genet. Nov 7;14(11):e1007749. PMID: 30403660
- Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. 2018. Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, Doench JG. Nat Commun. Dec 21;9(1):5416. PMID: 30575746
- Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. 2018. Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG. Nat Commun. Jun 27;9(1):2489. PMID: 29950558
2017
- A Broad-Spectrum Inhibitor of CRISPR-Cas9. 2017. Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, Kranzusch PJ, Sontheimer EJ, Davidson AR, Maxwell KL, Doudna JA. Cell. Sep 7;170(6):1224-1233.e15. PMID: 28844692
- Engineered Cpf1 variants with altered PAM specificities. 2017. Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F. Nat Biotechnol. 35(8):789-792. PMID: 28581492
- Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. 2017. Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, Sternberg SH, Joung JK, Yildiz A, Doudna JA. Nature. 550(7676):407-410. PMID: 28931002
- Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. 2017. Komor AC, Zhao KT, Packer MS, Gaudelli NM, Waterbury AL, Koblan LW, Kim YB, Badran AH, Liu DR. Sci Adv. 3(8):eaao4774. PMID: 28875174
- Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. 2017. Rees HA, Komor AC, Yeh WH, Caetano-Lopes J, Warman M, Edge ASB, Liu DR. Nat Commun. 8:15790. PMID: 28585549
- In Situ Capture of Chromatin Interactions by Biotinylated dCas9. 2017. Liu X, Zhang Y, Chen Y, Li M, Zhou F, Li K, Cao H, Ni M, Liu Y, Gu Z, Dickerson KE, Xie S, Hon GC, Xuan Z, Zhang MQ, Shao Z, Xu J. Cell. Aug 24;170(5):1028-1043.e19. PMID: 28841410
- Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. 2017. Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Nat Biotechnol. 35(4):371-376. PMID: 28191901
- Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. 2017. Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. Cell. Jan 12;168(1-2):150-158.e10. PMID: 28041849
- Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. 2017. Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F. Nat Biotechnol. 35(1):31-34. PMID: 27918548
- Nucleic acid detection with CRISPR-Cas13a/C2c2. 2017. Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ, Zhang F. Science. 356(6336):438-442. PMID: 28408723
- Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. 2017. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Nature. 551(7681):464-471. PMID: 29160308
- RNA editing with CRISPR-Cas13. 2017. Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. Science. 358(6366):1019-1027. PMID: 29070703
- RNA targeting with CRISPR-Cas13. 2017. Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, Lander ES, Voytas DF, Ting AY, Zhang F. Nature. 550(7675):280-284. PMID: 28976959
2016
- High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. 2016. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. Nature. 529(7587):490-5. PMID: 26735016
- Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. 2016. Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T. Nat Biotechnol.May;34(5):528-30. PMID: 27088723
- Naturally occurring off-switches for CRISPR-Cas9. 2016. Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, Davidson AR. Cell. 167(7):1829-1838. PMID: 27984730
- Practical Considerations for Using Pooled Lentiviral CRISPR Libraries. 2016. McDade JR, Waxmonsky NC, Swanson LE, Fan M. Curr Protoc Mol Biol. 115:31.5.1-31.5.13. PMID: 27366891
- Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. 2016. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Nature. 533(7603):420-4. PMID: 27096365
- Rationally engineered Cas9 nucleases with improved specificity. 2016. Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Science. 351(6268):84-8. PMID: 26628643
2015
- A Scalable Genome-Editing-Based Approach for Mapping Multiprotein Complexes in Human Cells. 2015. Dalvai M, Loehr J, Jacquet K, Huard CC, Roques C, Herst P, Côté J, Doyon Y. Cell Rep. 13(3):621-33. PMID: 26456817
- An updated evolutionary classification of CRISPR-Cas systems. 2015. Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. Nat Rev Microbiol. 13(11):722-36. PMID: 26411297
- Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. 2015. Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR. Nat Biotechnol. 33(1):73-80. PMID: 25357182
- CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. 2015. Savic D, Partridge EC, Newberry KM, Smith SB, Meadows SK, Roberts BS, Mackiewicz M, Mendenhall EM, Myers RM. Genome Res. 25(10):1581-9. PMID: 26355004
- Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. 2015. Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cell. 163(3):759-71. PMID: 26422227
- CRISPR-Cas9-Mediated Genetic Screening in Mice with Haploid Embryonic Stem Cells Carrying a Guide RNA Library. 2015. Zhong C, Yin Q, Xie Z, Bai M, Dong R, Tang W, Xing YH, Zhang H, Yang S, Chen LL, Bartolomei MS, Ferguson-Smith A, Li D, Yang L, Wu Y, Li J. Cell Stem Cell. 17(2):221-32. PMID: 26165924
- Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. 2015. Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Nat Biotechnol. 33(6):661-7. PMID: 25961408
- Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. 2015. Hashimoto M, Takemoto T. Sci Rep. 5:11315. PMID: 26066060
- Engineered CRISPR-Cas9 nucleases with altered PAM specificities. 2015. Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, Aryee MJ, Joung JK. Nature. 523(7561):481-5. PMID: 26098369
- Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. 2015. Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, Lim WA. Cell. 160(1-2):339-50. PMID: 25533786
- Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. 2015. Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Nat Biotechnol. 33(5):510-7. PMID: 25849900
- Genome engineering using CRISPR-Cas9 system. 2015. Cong L, Zhang F. Methods Mol Biol. 1239:197-217. PMID: 25408407
- Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. 2015. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang. Nature. 517(7536):583-8. PMID: 25494202
- High-throughput functional genomics using CRISPR-Cas9. 2015. Shalem O, Sanjana NE, Zhang F. Nat Rev Genetics. 16(5):299-311. PMID: 25854182
- Highly efficient Cas9-mediated transcriptional programming. 2015. Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM. Nat Methods. 12(4):326-8. PMID: 25730490
- In vivo genome editing using Staphylococcus aureus Cas9. 2015. Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. Nature. 520(7546):186-91. PMID: 25830891
- Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. 2015. Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Nat Biotechnol. 33(5):543-8. PMID: 25803306
- Multicolor CRISPR labeling of chromosomal loci in human cells. 2015. Ma H, Naseri A, Reyes-Gutierrez P, Wolfe SA, Zhang S, Pederson T. Proc Natl Acad Sci U S A. 112(10):3002-7. PMID: 25713381
- Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. 2015. Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, Davidson AR. Nature. 526(7571):136-9. PMID: 26416740
- Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. 2015. Shechner DM, Hacisuleyman E, Younger ST, Rinn JL. Nat Methods. 12(7):664-70. PMID: 26030444
2014
- A protein-tagging system for signal amplification in gene expression and fluorescence imaging. 2014. Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. Cell. 159(3):635-46. PMID: 25307933
- Crystal structure of Cas9 in complex with guide RNA and target DNA. 2014. Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Cell. 156(5):935-49. PMID: 24529477
- Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. 2014. Lin S, Staahl BT, Alla RK, Doudna JA. eLife. 3:e04766. PMID: 25497837
- Genetic Screens in Human Cells Using the CRISPR/Cas9 System. 2014. Wang T, Wei JJ, Sabatini DM, Lander ES. Science. 343(6166):80-4. PMID: 24336569
- Genome editing using Cas9 nickases. 2014. Trevino AE, Zhang F. Methods Enzymol. 546:161-74. PMID: 25398340
- Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. 2014. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Science. 343(6166):84-7. PMID: 24336571
- Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. 2014. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Cell. 159(3):647-6. PMID: 25307932
- Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. 2014. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Nat Biotechnol. 32(3):279-84. PMID: 24463574
- In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. 2014. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E, Lowe SW, Ventura A. Nature. 516(7531):423-7. PMID: 25337876
- Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. 2014. Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Nucleic Acids Res. 42(19):e147. PMID: 25122746
- Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. 2014. Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T.. Sci Rep. 4:5400. PMID: 24954249
- Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. 2014. Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Nat Biotechnol. 32(12):1262-7. PMID: 25184501
- Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. 2014. Anders C, Niewoehner O, Duerst A, Jinek M. Nature. 513(7519):569-73. PMID: 25079318
2013
- Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. 2013. Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Nature. 493(7432):429-32. PMID: 23242138
- CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. 2013. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. Nat Biotechnol. 31(9):833-8. PMID: 23907171
- CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. 2013. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. Cell. 154(2):442-51. PMID: 23849981
- DNA targeting specificity of RNA-guided Cas9 nucleases. 2013. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. Nat Biotechnol. 31(9):827-32. PMID: 23873081
- Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. 2013. Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Cell. 155(7):1479-91. PMID: 24360272
- Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. 2013. Fujita T, Fujii H. Biochem Biophys Res Commun. 439(1):132-6. PMID: 23942116
- Genome engineering using the CRISPR-Cas9 system. 2013. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Nat Protoc. 8(11):2281-308. PMID: 24157548
- High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. 2013. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. Nat Biotechnol. 31(9):822-6. PMID: 23792628
- Multiplex genome engineering using CRISPR/Cas systems. 2013. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Science. 339 (6121): 819–23. PMID: 23287718
- Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. 2013. Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R. Cell Res. 23(10):1163-71. PMID: 23979020
- One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. 2013. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. Cell. 153(4):910-8. PMID: 23643243
- Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. 2013. Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Cell. 152(5):1173-83. PMID: 23452860
- RNA-guided human genome engineering via Cas9. 2013. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. Science. 339(6121):823-6. PMID: 23287722